Schlüsselwörter: progressive hemifaziale Atrophie, Parry-Romberg-Syndrom
Progressive hemifaziale Atrophie – oder Parry-Romberg-Syndrom – ist eine seltene Krankheit, die zu den Formen der lokalisierten Morphea oder Sklerodermie gezählt wird. Ihre Ursache ist unbekannt. Sie ist durch eine Atrophie der Haut, des Fettgewebes, der Muskeln und der darunter liegenden osteokartilaginären Strukturen gekennzeichnet, die in der Regel einseitig Gesicht und Hals betrifft und mit neurologischen Symptomen (sekundäre Epilepsie) und der Beteiligung anderer Organe und Systeme verbunden ist. Der Verlauf ist langsam und progressiv und beginnt in den ersten beiden Lebensjahrzehnten. Es wurde eine Vorliebe für das weibliche Geschlecht beobachtet. Wir berichten über den Fall eines 10-jährigen Mädchens, das im Hipólito Unánue Hospital in Tacna, Peru, diagnostiziert wurde. Die Kenntnis dieser Erkrankung ist wichtig für die Differentialdiagnose von lokalisierten Morphen oder Sklerodermie.
Schlüsselideen
- Progressive hemifaziale Atrophie ist eine seltene Erkrankung mit einer Inzidenz von drei Fällen pro 100 000 Menschen pro Jahr; sie tritt häufiger bei Frauen auf und ihre Ätiologie ist unbekannt.
- Sie ist durch eine fortschreitende Atrophie gekennzeichnet, die in der Regel einseitig auf der linken Gesichtshälfte auftritt und die knöchernen und knorpeligen Strukturen beeinträchtigt.
- In dem berichteten Fall hatte der Patient die Läsion auf der rechten Seite, ein seltener Befund.
- Die Kenntnis dieser Krankheit ist wichtig für die Differentialdiagnose von Morphea oder lokalisierter Sklerodermie.
Einführung
Die progressive hemifaziale Atrophie, auch bekannt als Parry-Romberg-Syndrom, wurde erstmals von Caleb Hillier Parry und Moritiz Heinrich Romberg in den Jahren 1825 und 1846 beschrieben und ist durch eine fortschreitende Atrophie der Haut und des darunter liegenden Gewebes gekennzeichnet, die das Gesicht und den Hals meist einseitig betrifft. Die Inzidenz liegt bei etwa drei Fällen pro 100 000 Menschen und Jahr. Obwohl diese seltene Krankheit in der Regel nur einseitig Gesicht und Hals betrifft, tritt sie gelegentlich auch beidseitig auf und betrifft sogar den Rumpf, die Arme und die Beine. Der Verlauf ist progressiv, langsam und tritt hauptsächlich im Alter zwischen zwei und 20 Jahren auf.
Die Pathogenese dieser Krankheit ist nicht bekannt; es wurden jedoch eine Reihe von Theorien vorgeschlagen, darunter immunologische, traumatische, infektiöse, endokrinologische, neurologische und genetische Ursachen. Epilepsie ist die häufigste mit dem Perry-Romberg-Syndrom assoziierte Anomalie.
Wir berichten über ein 10-jähriges Mädchen, bei dem 2015 im Hipolito Unanue Hospital in Tacna, Peru, eine progressive hemifaziale Atrophie diagnostiziert wurde. Soweit wir wissen, ist dies der dritte in Peru gemeldete Fall und der erste in Tacna.
Fallbericht
Dies ist der Fall einer 10-jährigen Patientin aus Tacna (einer Stadt im Süden Perus an der Grenze zu Bolivien und Chile). Sie ist das Kind einer zweiten Trächtigkeit und wurde per Kaiserschnitt zur Welt gebracht. Ihre Eltern sind nicht blutsverwandt und ihre unmittelbaren Familienangehörigen sind vermutlich gesund. In der Perinatalperiode wurde keine relevante Krankheit festgestellt. Die körperliche und motorische Entwicklung war angemessen.
Unsere Patientin kam mit Bewusstseinsverlust und tonisch-klonischen Anfällen, die sich zu einem fokalen epileptischen Zustand entwickelten, in die Notaufnahme des Hipólito Unánue Krankenhauses. Aus diesem Grund wurde das Mädchen für weitere Untersuchungen und Behandlungen aufgenommen.
Bei der körperlichen Untersuchung stellten wir zwei atrophische Läsionen im Gesicht fest: Die erste und längste Läsion, die sich an der medialen Gesichtslinie befand, war acht Zentimeter lang, zwei Zentimeter breit und zwischen einem und zwei Millimeter tief und erstreckte sich vom Trichion bis zum rechten Nasenflügel; an ihrem Scheitelpunkt wurde eine leichte Hyperpigmentierung festgestellt (Abbildungen 1 und 2, gekennzeichnet durch einen roten Pfeil). Die zweite atrophische Läsion, die sich seitlich von der ersten Läsion befand, erstreckte sich vom rechten Stirnhaaransatz bis zum rechten Augenbrauenbogen, wo eine Zone der Madarose beobachtet wurde, und war vier Zentimeter lang, einen Zentimeter breit und sieben Millimeter tief (Abbildungen 1 und 2, Läsion durch grünen Pfeil gekennzeichnet).
Abbildung 1. Atrophische Läsionen im Gesicht.
Abbildung 2. Zone der Madarose.
In der rechten Parietalregion wurde eine Alopeziezone gefunden (Abbildung 3). Gleichzeitig beobachteten wir eine Anisokorie und Mydriasis der rechten Pupille, die jedoch immer noch photoreaktiv war. Außerdem wurden eine mäßige Ptosis rechts, ein Hornhautulkus rechts, eine Dislokation der rechten Linse und eine Ametropie festgestellt. An der Netzhaut wurden keine Abnormitäten festgestellt. Es wurde eine Atrophie des rechten Nasenflügels festgestellt, zusammen mit einer Septumdeviation, einer erhöhten rechten Mundpartie, einer Akzentuierung des rechten nasogenen Sulcus (Abbildung 2, gelber Pfeil) und einer abnormen Zahnstellung (Abbildung 4). Der Rest der Untersuchung war normal.
Abbildung 3. Alopecische Zone im rechten Scheitelbereich.
Abbildung 4. Abnorme Zahnstellung.
Im Alter von drei Jahren erlitt unser Patient ein Hirntrauma mit Bewusstseinsverlust und Krampfanfällen. Damals wurde angenommen, dass diese Symptome durch das Trauma verursacht wurden. Ein Jahr später bemerkten die Eltern der Patientin das Vorhandensein und das Fortschreiten der Gesichtsanomalie (wie oben beschrieben); sie suchten jedoch keine ärztliche Hilfe, weil sie annahmen, dass dies eine Folge des früheren Traumas war.
Die Laboruntersuchungen, die wir bei der Patientin durchführten, zeigten eine leichte Leukozytose mit Neutrophilie (18,3 x 103 Zellen pro mm3, 79 % Neutrophile) und eine erhöhte Glykämie (149,8 mg/dl). Diese Befunde wurden durch Krampfanfälle erklärt. Die Tests auf antinukleäre Antikörper, antineutrophile zytoplasmatische Antikörper, Hepatitis-B-Oberflächenantigen und IgM gegen das Hepatitis-A-Virus waren negativ (normal).
Die Computertomographie (CT) des Gehirns zeigte Verkalkungen und ein umgebendes Ödem in den rechten Basalganglien: im Putamen und im Kopf des Nucleus caudatus (Abbildung 5). In der 3D-CT-Rekonstruktion des Schädels fanden wir eine Asymmetrie der Gesichtsknochen: Atrophie des rechten Oberkieferknochens und des Stirnbeins, anormale Zahnstellung, Abweichung der Nasenscheidewand und Überwuchs der unteren linken Nasenmuschel (Abbildung 6).
Abbildung 5. Verkalkungen in den Basalganglien des Gehirns.
Abbildung 6. Nasenseptumdeviation mit Linkskonvexität.
Nachdem wir die Daten aus der Anamnese, der körperlichen Untersuchung und den diagnostischen Tests erhalten hatten, führte der Hauptautor dieses Artikels eine umfassende Suche in Datenbanken für seltene Krankheiten durch (z. B. Mendelian Inheritance Online in Man – OMIM, verfügbar unter www.omim.org) und kam zu dem Schluss, dass der Zustand des Mädchens mit dem Parry-Romberg-Syndrom oder der progressiven hemifazialen Atrophie vereinbar war.
Nachdem eine angemessene Kontrolle der Anfälle erreicht worden war, wurde die Patientin aus dem Krankenhaus entlassen; sie steht jedoch unter ärztlicher Beobachtung durch einen Neurologen, einen Kinderarzt, einen Augenarzt und einen Psychologen. Dieses multidisziplinäre Team hat das Ziel, die Lebensqualität unserer Patientin zu verbessern.
Diskussion
Das Parry-Romberg-Syndrom ist eine seltene Krankheit, die durch eine fortschreitende Atrophie der Haut, der Muskeln und der Knochen gekennzeichnet ist, die das Gesicht beeinträchtigt, in der Regel einseitig, und in einigen Fällen von Anomalien des Gebisses, der Zunge und des Gaumens begleitet wird. Obwohl das Parry-Romberg-Syndrom in der Regel einseitig ist, können auch Patienten mit beidseitigen Schäden gefunden werden, sogar solche mit beeinträchtigtem Rumpf, Armen und Beinen.
Die Inzidenz dieser Krankheit liegt bei drei Fällen pro 100 000 Patienten pro Jahr und ist bei Frauen mit einem Verhältnis von 3:1 häufiger. Sie tritt typischerweise in den ersten zwanzig Lebensjahren auf.
Diese Krankheit wird gewöhnlich als lineare lokalisierte Esklerodermie oder Morphea klassifiziert. In der Weltliteratur fanden wir Hinweise auf das Parry-Romberg-Syndrom als Morphea in coup de sabre („Säbelhieb“, wegen der Ähnlichkeit der Hautatrophie mit der durch den Säbel verursachten Wunde). Nach unserem Verständnis handelt es sich jedoch um zwei unterschiedliche Erkrankungen, da die Patienten mit Parry-Romberg-Syndrom keine histologischen Merkmale der Hautsklerose aufweisen, während Patienten mit Morphea in coup de sabre diese aufweisen.
Die Ätiologie des Parry-Romberg-Syndroms ist noch unbekannt; die am meisten akzeptierte Hypothese ist jedoch die Autoimmun-Ätiologie. Die Th1- und Th17-Entzündungswege sind für die Pathophysiologie dieser Krankheit, zumindest in den frühen Stadien, von großer Bedeutung. Der Th2-Entzündungsweg ist im Spätstadium, dem fibrotischen Stadium, von Bedeutung. Diese Entzündungsreaktion des Gewebes könnte durch ein zufälliges traumatisches Ereignis, eine Operation oder sogar ein geburtshilfliches Trauma ausgelöst werden. Die neurologische Theorie besagt, dass diese Krankheit das Ergebnis einer desorganisierten Migration von Neuralleistenzellen ist, die einen oder mehrere Äste des Trigeminusnervs beeinträchtigt.
Einigen Berichten zufolge ist das Parry-Romberg-Syndrom eine genetisch autosomal dominante Krankheit. Außerdem wird behauptet, dass die progressive hemifaziale Atrophie mit Borreliose, Borrelia burgdorferi und anderen Infektionen wie Syphilis, Röteln, Tuberkulose und dem Hepatitis-B-Virus in Verbindung steht.
In diesem Fall entwickelte der Patient die Krankheit nach einem Sturz, bei dem er ein Kopftrauma erlitt. In der Familie gab es keine Vorgeschichte von Krankheiten. Ihre Labortests auf antinukleäre Antikörper und virale Hepatitis waren negativ. Wir haben unsere Patientin nicht auf Borreliose untersucht, da Peru kein endemisches Gebiet für diese Krankheit ist und sie bestritt, in endemische Regionen gereist zu sein.
Das Parry-Romberg-Syndrom betrifft Dermatome eines oder mehrerer Äste des Nervus trigeminus und beeinträchtigt gewöhnlich die linke Gesichtshälfte. Die Pigmentierung und Helligkeit der Haut kann in gewissem Maße zunehmen. Alopezie kann in jedem Bereich der Kopfhaut auftreten, in der Regel jedoch in der frontoparietalen Region.
M. Wong zeigt in seiner Fallserie eine Krankheitsvorliebe für die linke Gesichtshälfte. Carlos Galarza zeigte in seinen beiden aus Cuzco, Peru, berichteten Fällen ebenfalls eine Hautatrophie auf der linken Seite. Unser Patient wies eine Hautatrophie in der rechten Gesichtshälfte auf, die sich von der Frontal- bis zur Unterkieferregion erstreckte und von einer leichten Hyperpigmentierung, einer Septumdeviation, einer abnormalen Verteilung der Zähne und einer parietalen Alopezie begleitet wurde.
Augenmanifestationen des Parry-Romberg-Syndroms können vor, während oder nach dem Auftreten der Hautatrophie auftreten und umfassen: Enophthalmus, der zu Diplopie führt (verursacht durch Fettatrophie im Retro-Bulbus-Raum), trockenes Auge oder Uveitis. Es wurde auch über Lidpigmentierung, Photophobie, Schielen, Irisatrophie, Panuveitis, hypotone Ziliarkörper, Netzhautvaskulitis, Netzhautödeme und -ablösungen usw. berichtet.
Die neurologische Untersuchung kann eine Augenmuskellähmung mit Pupillenanomalien, Anisokorie, Mydriasis oder Myosis auf der betroffenen Seite und eine Beteiligung des Sehnervs zeigen, die zu Neuroretinitis und Papillitis führen kann. Unsere Patientin litt seit ihrem ersten Besuch in der Notaufnahme an einer schweren Anisokorie und Mydriasis des rechten Auges, einer Anomalie der Pupillenreaktion, und mit dem Fortschreiten der Krankheit traten weitere Manifestationen auf: Dislokation der rechten Linse, Hornhautgeschwür, schwerer Enophthalmus und Ametropie.
Dalla Costa berichtete in ihrer weltweiten Untersuchung von 205 Patienten, bei denen das Parry-Romberg-Syndrom diagnostiziert wurde, dass 50 % der Patienten Symptome eines beeinträchtigten zentralen Nervensystems aufwiesen und 15 % epileptische Anfälle, Kopfschmerzen, Gesichtsschmerzen, Hirnnervenbeteiligung und Halbseitenlähmung hatten.
Die häufigste neurologische Manifestation des Parry-Romberg-Syndroms ist die Epilepsie (60,5 % Häufigkeit). Fokale Anfälle ipsilateral zu Hirnverkalkungen sind häufig (in 50 % aller Fälle). Mit einer Häufigkeit von 33% ist die sekundäre Epilepsie sehr schwer zu behandeln. Kopfschmerzen (44%) und Trigeminusneuralgie (8,5%) können ebenfalls auftreten.
Intraparenchymale Hirnverkalkungen auf derselben Seite wie die Gesichtsanomalien sind die häufigsten Befunde in der Computertomographie und der Magnetresonanztomographie. Hyperintensitäten der weißen Substanz, fokale Infraktion des Corpus callosum, zerebrale Hemiatrophie und leptomeningeale Anreicherung, intrakranielle Aneurysmen und vaskuläre Fehlbildungen sowie Mikroblutungen im Gehirn wurden beschrieben. Es gibt keine Korrelation zwischen Neuroimaging und Gesichtsmerkmalen dieser Krankheit.
Obwohl neurologische und okuläre Merkmale die häufigsten Manifestationen des Parry-Romberg-Syndroms sind, können auch kardiale Anomalien (hypertrophe Kardiomyopathie), endokrine Anomalien (Hyperthyreose, Hypothyreose), Autoimmunerkrankungen (primäre biliäre Zirrhose, rheumatoide Arthritis, multiple Sklerose) und angeborene Erkrankungen wie das Poland-Syndrom, Mikrophthalmie und Nierenfehlbildungen auftreten.
Sekundäre Epilepsie und Kopfschmerzen waren die vorherrschenden neurologischen Merkmale in unserem Fall. Die epileptischen Anfälle waren gut kontrolliert worden. Die Tomographie- und Resonanzergebnisse zeigten Verkalkungen in den Basalganglien: Putamen und Nucleus caudatus. Wir fanden keine anderen Anomalien als die oben beschriebenen.
Das Ziel der Behandlung ist es, die aktive Phase der Krankheit zu stoppen. Dies kann mit der Verschreibung von Methotrexat in einer Dosierung von 0,3 – 1mg/kg/Woche für ein bis zwei Jahre erreicht werden, und es kann in den ersten drei Monaten mit Prednison kombiniert werden, wenn man die verzögerte Wirkung von Methotrexat auf Entzündung und Fibrose berücksichtigt.
Das Parry-Romberg-Syndrom hat aufgrund der funktionellen und ästhetischen Einschränkungen einen großen biopsychosozialen Einfluss auf das Leben der Patienten. Es wurden einige chirurgische Ansätze zur Wiederherstellung der Gesichtssymmetrie vorgeschlagen. Leichte und mittelschwere Fälle können mit Silikonfüllern, Kollagen, porösen Polyethylenimplantaten und Eigenfetttransplantationen behandelt werden. Knorpel-, Knochen-, Fett- und Hauttransplantationen sind Behandlungsoptionen für schwerere Fälle. Das Ziel der psychologischen Behandlung ist die soziale Wiedereingliederung der Patienten.
Die Prognose der Krankheit hängt vom Alter des Patienten ab, wobei sie schwerer ist, wenn die atrophischen Veränderungen im Erwachsenenalter (ab 20 Jahren) beginnen. Der Schweregrad der Atrophie, die Hirnschädigung und das schlechte Ansprechen auf die Behandlung sind bei Patienten, die älter als 20 Jahre sind, höher. Diese Determinanten geben uns einen globalen Ansatz für die Folgeerscheinungen und die Behandlung, die in Betracht gezogen werden muss, um bessere therapeutische Ergebnisse zu erzielen.
Schlussfolgerungen
Die progressive Gesichtsatrophie oder das Parry-Romberg-Syndrom ist eine seltene, fortschreitende Krankheit unbekannter Ursache, die hauptsächlich Frauen betrifft. Es gibt nur wenige Fälle dieses Syndroms in Peru, und dies ist der erste Fall in der Region Tacna.
Es ist durch das Fortschreiten der Atrophie gekennzeichnet, die in der Regel einseitig ist, häufiger auf der linken Seite des Gesichts auftritt und osteokartilaginöse Strukturen betrifft. In unserem Fall befand sich die Hautanomalie auf der rechten Seite; dies ist ein seltener Befund.
Die Kenntnis dieser Krankheit wird bei der Differentialdiagnose von atrophischen Läsionen auf einer Gesichtsseite relevant. Unter den Krankheiten, bei denen eine Differentialdiagnose gestellt werden muss, sind folgende hervorzuheben: Säbelzahnkrankheit, tiefer Lupus, Steroidatrophie und andere Arten von Morphea.
Beiträge der Autoren
EOL: Hauptautor, Diagnose und Nachsorge des Patienten, Verfassen und kritische Überprüfung des Artikels, endgültige Genehmigung des Artikels und Übernahme der Verantwortung für das Manuskript. SDA: Autor, Verfassen und wichtige Beiträge zum Artikel und Übernahme der Verantwortung für das Manuskript. PDA: Co-Autor, wichtige Beiträge zum Artikel
Interessenkonflikt
Keiner der Autoren erklärt, Interessenkonflikte mit dem Thema dieses Artikels zu haben.
Finanzierungsquelle
Die Finanzierung bei der Ausarbeitung des Artikels ist eigene.
Ethische Aspekte
Die Autoren haben die informierte Zustimmung der Eltern der Patientin (da sie minderjährig ist) für die Veröffentlichung dieses Artikels und der dazugehörigen Bilder eingeholt.
Daten
Wir erklären, dass wir auf Anfrage Daten zur Verfügung stellen können.
Von den Herausgebern
Die ursprüngliche Version dieses Manuskripts wurde in Spanisch eingereicht. Diese englische Version wurde von den Autoren eingereicht und von der Zeitschrift leicht redigiert.

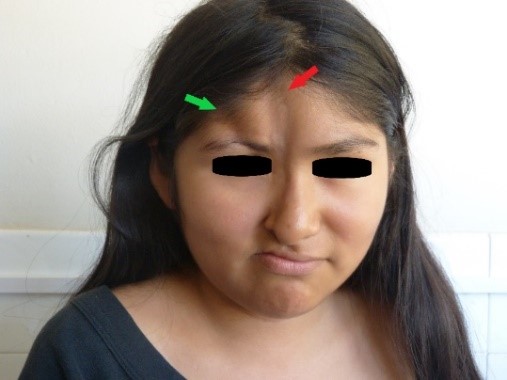 Abbildung 1. Atrophische Läsionen im Gesicht.
Abbildung 1. Atrophische Läsionen im Gesicht. 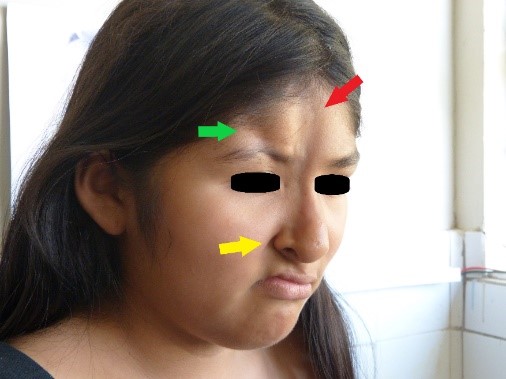 Abbildung 2. Zone der Madarose.
Abbildung 2. Zone der Madarose. 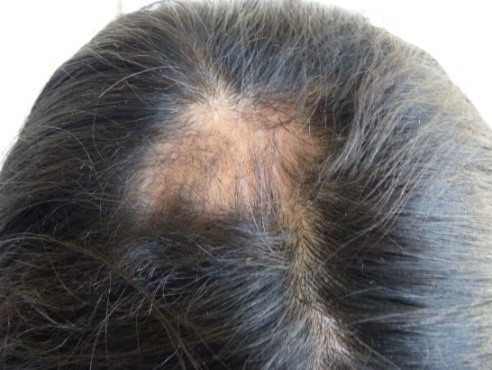 Abbildung 3. Alopezische Zone in der rechten Parietalregion.
Abbildung 3. Alopezische Zone in der rechten Parietalregion. 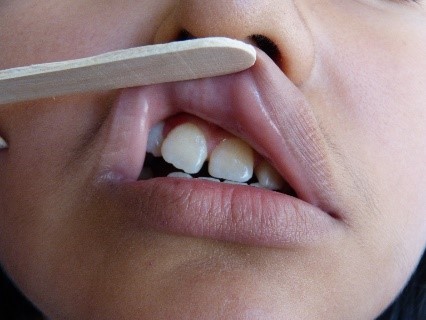 Abbildung 4. Abnorme Stellung der Zähne.
Abbildung 4. Abnorme Stellung der Zähne. 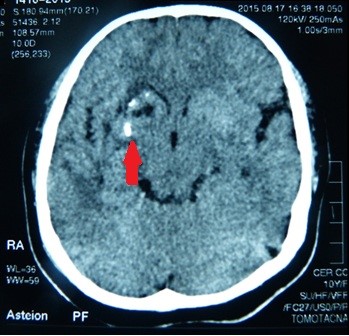 Abbildung 5. Verkalkungen in den Basalganglien des Gehirns.
Abbildung 5. Verkalkungen in den Basalganglien des Gehirns. 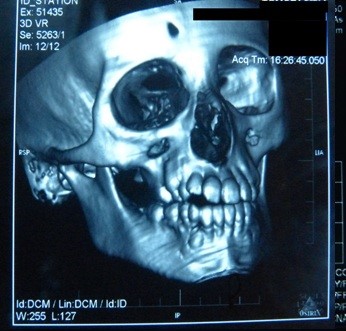 Abbildung 6. Nasenscheidewandabweichung mit Linkskonvexität.
Abbildung 6. Nasenscheidewandabweichung mit Linkskonvexität.