Die chronische traumatische Enzephalopathie (CTE) ist eine ausgeprägte, mit Tau-Protein assoziierte neurodegenerative Erkrankung. Die CTE-Diagnose wurde vermehrt bei Sportlern, insbesondere American-Football-Spielern, sowie bei Militärveteranen in Kampfsituationen gestellt (1, 2). Obwohl CTE erst seit relativ kurzer Zeit öffentlich anerkannt ist, wurde es erstmals in einem klassischen Artikel von Martland et al. (3) als „punch-drunk“-Syndrom beschrieben. Der Bericht konzentrierte sich auf eine Reihe von Boxern, die während ihrer Karriere wiederholt Kopfschläge erlitten hatten und sowohl psychiatrische Symptome als auch schwere Gedächtnisstörungen und neurokognitive Defizite aufwiesen, die denen typischer Demenzpatienten ähnelten (3). Die Krankheitsbezeichnung entwickelte sich zu „dementia pugilistica“ (4) und schließlich 1949 zu CTE (5).
CTE weist ein einzigartiges neuropathologisches Merkmal auf, das aus einer Anhäufung von phosphoryliertem Tau (p-tau) in Sulci und perivaskulären Regionen, Mikrogliosis und Astrozytose besteht. Diese pathologischen Veränderungen führen zu einer fortschreitenden, schwächenden Neurodegeneration. Auf der Grundlage des Musters der pathologischen Progression wird die CTE in vier Stadien unterteilt (Abbildung 1). Im Stadium I der CTE erscheint das Gehirn grob gesehen normal, aber p-tau wird an einer begrenzten Anzahl von Stellen gefunden, häufig in den lateralen und frontalen Kortizes sowie proximal von kleinen Blutgefäßen in der Tiefe der Sulci. Es kann eine geringe Anzahl von neurofibrillären Tangles (NFTs) und Neuriten im Locus coeruleus vorhanden sein. Im Stadium II können lokalisierte makroskopische Anomalien festgestellt werden. Bei groben anatomischen Schnitten und in der Neurobildgebung werden eine Vergrößerung der Seitenventrikel, ein Cavum septum pellucidum mit oder ohne Fenestration sowie eine Blässe des Locus coeruleus und der Substantia nigra beobachtet. Es gibt mehrere Herde von p-tau in der Tiefe der Sulci, und es gibt ein sich ausbreitendes Muster. Im Stadium III zeigen die meisten groben pathologischen Schnitte makroskopische Anomalien. Es besteht ein globaler Gewichtsverlust des Gehirns, eine leichte Atrophie der Frontal- und Temporallappen und eine Erweiterung der Ventrikel. Die Hälfte der CTE-Patienten weist Septumanomalien auf, einschließlich Cavum septum pellucidum. Die P-Tau-Pathologie breitet sich aus und betrifft den frontalen, temporalen, parietalen und insulären Kortex. Im Stadium IV ist die Gewichtsabnahme des Gehirns dramatisch, und es wurde von einem Gehirngewicht von 1.000 g berichtet (im Vergleich zu 1.300-1.400 g bei normalen Gehirnen). Es besteht eine tiefgreifende Atrophie des Frontal- und des medialen Temporallappens sowie der vorderen Thalami. Außerdem kommt es zu einer Atrophie der weißen Substanzbahnen. Die Mehrzahl der Patienten im vierten Stadium weist Septumanomalien auf. Die Ausbreitung des p-tau betrifft die meisten Regionen, darunter auch den kalzarinen Kortex (7, 8). Anomalien des phosphorylierten 43 kDa TAR-DNA-Bindungsproteins (TDP-43) sind bei den meisten CTE-Patienten ebenfalls zu beobachten. Die parenchymale TDP-43-Pathologie ist ebenfalls progressiv, ähnlich wie das anatomische Muster der Ausbreitung von p-tau. TDP-43-Immunreaktivität findet sich in fast allen Fällen einer Erkrankung im Stadium IV (7).

Abbildung 1. Die obigen Abbildungen stellen die vier Stadien der CTE nach McKee dar.
Der klinische Phänotyp der CTE ist noch nicht eindeutig definiert. In den folgenden Abschnitten werden Versuche zur Charakterisierung der CTE-Symptome in den verschiedenen Stadien des Krankheitsprozesses skizziert (Tabelle 1). Nach der Klassifizierung von McKee ist ein typischer CTE-Patient im Stadium I asymptomatisch oder klagt über leichte Defizite des Kurzzeitgedächtnisses und depressive Symptome. Leichte Aggressionen können beobachtet werden. In Stadium II können die Stimmungs- und Verhaltenssymptome Verhaltensausbrüche und schwerere depressive Symptome umfassen. In Stadium III weisen die Patienten typischerweise vermehrt kognitive Defizite auf, darunter Gedächtnisverlust, Defizite bei den exekutiven Funktionen, Störungen des räumlichen Vorstellungsvermögens und Apathie. Im Stadium IV weisen die Patienten fortgeschrittene Sprachdefizite, psychotische Symptome einschließlich Paranoia, motorische Defizite und Parkinsonismus auf.
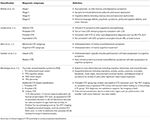
Tabelle 1. Vorgeschlagene klinische Klassifizierungen von CTE.
Jordan et al. (10) waren eine der ersten, die die Krankheit klinisch charakterisierten. Sie unterteilten die klinischen Präsentationen von CTE in drei Bereiche: Verhalten/Psychiatrie, Kognition und Motorik. Der verhaltensbezogene und psychiatrische Bereich umfasste Aggression, Depression, Apathie, Impulsivität, Wahnvorstellungen einschließlich Paranoia und Suizidalität. Der kognitive Bereich umfasste verminderte Aufmerksamkeit und Konzentration, Gedächtnisdefizite, Defizite in der Exekutivfunktion, visuell-räumliche Störungen, Sprachdefizite und Demenz. Die motorischen Merkmale schließlich umfassten Dysarthrie, Gangstörungen, Ataxie und Koordinationsschwäche, Spastizität und Parkinsonismusmerkmale wie Zittern. Auf der Grundlage dieser klinischen Merkmale sowie vorhandener neuropathologischer Informationen wurden vier diagnostische Subtypen definiert, nämlich „definitiver“, „wahrscheinlicher“, „möglicher“ und „unwahrscheinlicher“ CTE.
Stern et al. (11) und verwandte Fallberichte (14, 15) unterschieden sich in ihrer Beschreibung eines typischen CTE-Patienten, indem sie die klinische Präsentation in zwei verschiedene Subtypen konzeptualisierten. Der erste Subtyp wies hauptsächlich Verhaltens- und Stimmungsänderungen auf, während der andere hauptsächlich kognitive Beeinträchtigungen zeigte. Die überwiegende Mehrheit des Subtyps mit Stimmungs-/Verhaltensveränderungen entwickelte mit fortschreitender Krankheit kognitive Defizite. Allerdings zeigten relativ wenige Patienten der kognitiven Gruppe während des Krankheitsverlaufs Stimmungs- oder Verhaltensveränderungen. In der Studie von Stern et al. (11) hatten die Patienten der kognitiven Gruppe eine deutlich höhere Wahrscheinlichkeit, eine Demenz zu entwickeln. Außerdem waren sie zum Zeitpunkt der Diagnose deutlich älter als die Patienten der Stimmungs-/Verhaltensgruppe. Die verhaltensbezogene Untergruppe der CTE-Patienten kann Patienten ähneln, die an einer verhaltensbezogenen Variante der frontotemporalen Demenz (bvFTD) leiden, was die klinische Diagnose erschwert. Allerdings sind die für die bvFTD typischen Verhaltensmanifestationen wie Apathie und Enthemmung bei CTE-Patienten oft nicht zu beobachten (11, 16). In Anbetracht der inhärenten Heterogenität der bvFTD sowie der ähnlichen tauopathischen Natur beider Krankheiten stellt die Unterscheidung zwischen bvFTD und CTE eine diagnostische Herausforderung dar.
Unter den Verhaltenssymptomen der CTE bleibt die Assoziation zwischen Suizid und CTE ein Thema, das in der Literatur untersucht wird. Frühere Studien, wie die von Omalu et al. (17) berichtete Serie von fünf Profisportlern mit bestätigter CTE-Diagnose, hatten einen engen Zusammenhang zwischen CTE und Selbstmord nahegelegt. Die Autoren schlugen ferner vor, dass die Ätiologie von suizidalem/parasuizidalem Verhalten in der CTE-Population teilweise auf Tauopathie in Form von neurofibrillären Tangles und neuritischen Fäden in strategischen limbischen Gehirnkernen wie dem Locus ceruleus zurückzuführen sein könnte. Maroon et al. (18) untersuchten 153 pathologisch bestätigte Fälle von CTE, die zwischen 1954 und 2013 veröffentlicht wurden. Sie berichteten, dass die Suizidprävalenz in der CTE-Population und bei den Unfalltoten 11,7 bzw. 17,5 % beträgt und damit deutlich über den Werten der Allgemeinbevölkerung von 1,5 bzw. 4,8 % liegt (18). Befürworter der gegenteiligen Ansicht weisen darauf hin, dass Selbstmorde vor allem in früheren Stadien der CTE gemeldet wurden und der Zusammenhang zwischen Krankheitsverlauf und Selbstmord derzeit noch unklar ist (19).
In einer Meta-Analyse von 158 Fallstudien durch Gardner et al. (12) wurden die klinischen CTE-Symptome in „klassische“ und „moderne“ CTE-Symptome unterteilt, um eine Unterscheidung zwischen einer älteren Beschreibung von CTE-Fällen, die sich vor allem auf Boxer konzentrierte, und einer weiterentwickelten klinischen Beschreibung zu treffen, die auch für professionelle American-Football-Spieler gilt. Während zu den „klassischen“ CTE-Symptomen typischerweise Dysarthrie, Bewegungsstörungen und später auch Gedächtnisstörungen gehören, umfasst das „moderne“ CTE-Bild auch neuropsychiatrische Symptome wie depressive Symptome, Paranoia, sozialer Rückzug und Isolation, beeinträchtigtes Urteilsvermögen und Aggression. Kognitive Defizite wie Gedächtnisverlust, exekutive Dysfunktion, Sprach- und Informationsverarbeitungsdefizite treten erst im späteren Verlauf des Krankheitsprozesses auf (12).
Da die Definition von CTE in erster Linie von pathologischen Merkmalen abhängt, wurde von Montenigro et al. (13) ein alternativer klinischer Begriff für das traumatische Enzephalopathie-Syndrom (TES) vorgeschlagen, der die klinischen Folgen wiederholter Schädel-Hirn-Traumata beschreibt. Die Autoren stützten diese Klassifizierung auf eine Überprüfung von 202 veröffentlichten Fällen. TES ist eine umfassendere Diagnose und kann in vier Unterkategorien unterteilt werden, darunter TES Verhaltens-/Stimmungsvariante, TES kognitive Variante, TES gemischte Variante und TES Demenz. Die vorgeschlagene TES-Diagnose basiert auf dem Vorhandensein von fünf allgemeinen Kriterien, drei klinischen Kernmerkmalen und neun unterstützenden Merkmalen. Unter Verwendung vorhandener Biomarker* (Tabelle 1) wurden zusätzliche diagnostische Qualifizierungen vorgeschlagen, darunter „wahrscheinliche“, „mögliche“ und „unwahrscheinliche“ CTE (9, 13). Die vorgeschlagene TES-Diagnose enthielt auch zeitliche Qualifizierungsmerkmale und umfasste „progressiver Verlauf“, „stabiler Verlauf“ und „unbekannter/inkonsistenter Verlauf“. Wenn die klinische Präsentation auch motorische Anzeichen wie Parkinsonismus enthielt, wurde der Modifikator „mit motorischen Merkmalen“ hinzugefügt.
Während unser Verständnis von CTE wächst, gibt es eine Reihe von Herausforderungen und Kritikpunkten, die angegangen werden müssen. Eine Hypothese, die eine Alternative zum Phänomen CTE darstellt, ist die Theorie einer verminderten „kognitiven Reserve“. Diese Theorie besagt, dass wiederholte Neurotraumata zu einer Verringerung der kognitiven Reserve führen und die Entwicklung einer zugrunde liegenden neurodegenerativen Störung beschleunigen (20, 21). Wenn diese Theorie zutrifft, würde dies bedeuten, dass CTE und Alzheimer auf demselben neuropathologischen Spektrum liegen. Diese Behauptung verdient eine weitere Analyse. Ähnlich wie bei der Alzheimer-Krankheit bestehen auch die Tau-Isoformen bei der CTE aus einer Mischung von Isoformen mit drei Wiederholungen (3R) und vier Wiederholungen (4R). Einem kürzlich erschienenen Bericht von Falcon et al. (22) zufolge enthalten die aus den Gehirnen von CTE-Patienten extrahierten Tau-Filamente jedoch auch eine einzigartige ß-Helix-Region mit einem hydrophoben Hohlraum, die in den Gehirnen von AD-Patienten nicht vorhanden ist. Der Hohlraum enthält einen zusätzlichen Kofaktor, von dem angenommen wird, dass er eine funktionelle Rolle bei der Tau-Ausbreitung spielt. Falcon et al. (22) vermuten, dass die Lage der Tau-Einschlüsse in der Nähe von Blutgefäßen darauf hindeutet, dass Kofaktoren, die für die Tau-Assemblierung erforderlich sind, nach einem Kopftrauma die Blut-Hirn-Schranke passieren können. Die Autoren argumentieren weiter, dass die Tatsache, dass ein Hirntrauma nur bei einer Untergruppe der verletzten Bevölkerung zu CTE führt, mit einem höheren Gehalt an Kofaktoren bei den anfälligeren Personen zusammenhängen könnte. Diese Kofaktoren könnten ein therapeutisches Ziel für die Verhinderung der Tau-Assemblierung und der Entwicklung von CTE nach einer Verletzung darstellen (22).
Eine alternative Theorie besagt, dass die bei CTE-Patienten berichteten psychiatrischen Symptome wie Depression und Wut unabhängig vom CTE-Krankheitsprozess sind und auf eine gemeinsame Grundlage zurückgeführt werden. Die Befürworter dieser Hypothese berufen sich auf frühere Studien wie die von Weir et al. (23), in der 1.063 ehemalige NFL-Spieler befragt wurden, ob sie Wutanfälle erlebt haben. Es wurde berichtet, dass 30,7 % der Spieler im Alter von 30 bis 49 Jahren und 29,3 % der Spieler im Alter von 50 Jahren oder darüber über Wutausbrüche berichteten. Die Autoren wiesen jedoch auch darauf hin, dass die gemeldeten Wutausbrüche in der Tat niedriger waren als in der allgemeinen US-Bevölkerung, nämlich 54,8 % bei Männern zwischen 30 und 49 Jahren und 47,2 % bei Männern über 50 Jahren (23). Obwohl die Argumente bezüglich der Komorbidität von psychiatrischen Symptomen und neurodegenerativen Erkrankungen wie CTE auf der Grundlage von Neurobildgebung und neuropathologischen Befunden schwer zu verifizieren sind, kann man ähnliche Argumente auf psychiatrische Symptome bei allen neurodegenerativen Erkrankungen wie Alzheimer, bvFTD, Parkinson oder amyotrophe Lateralsklerose (ALS) anwenden.
Eine weitere wichtige Quelle für diagnostische Verwirrung ist die klinische Abgrenzung zwischen CTE und dem verlängerten postkonkussiven Syndrom (PCS), insbesondere angesichts früherer Berichte, die darauf hinweisen, dass ~10-20 % der Personen, die eine Gehirnerschütterung erleiden, verlängerte Symptome aufweisen. Das chronische postkonkussive Syndrom (CPCS) bezeichnet das Fortbestehen von PCS-Symptomen, die zu einer Beeinträchtigung der funktionellen und häufig auch der sportlichen Leistungsfähigkeit führen und länger als ein Jahr andauern. Zu den CPCS-Symptomen gehören Kopfschmerzen, Schwindel, Beeinträchtigungen der Aufmerksamkeit, des Gedächtnisses und der exekutiven Funktionen sowie Depressionen und Reizbarkeit (10). King und Kirkwilliam prägten den Begriff „Permanent PCS“, um diejenigen zu bezeichnen, bei denen die PCS-Symptome durchschnittlich 6,9 Jahre nach der ersten Gehirnerschütterung fortbestehen. Darüber hinaus berichteten sie, dass eine signifikante Anzahl von Patienten mit permanenter PCS (40-59 %) auch prämorbide oder postmorbide neuropsychiatrische Erkrankungen wie Depressionen, Angstzustände, PTBS und/oder Schmerzen aufwiesen (24). Wie von Jordan et al. (10) dargelegt, ist CPCS klinisch von CTE zu unterscheiden, und zwar aufgrund des zeitlichen Zusammenhangs mit dem akuten Erschütterungsereignis. Eine gründliche und genaue zeitliche Anamnese bleibt der Schlüssel zur neurologischen Beurteilung. Darüber hinaus sind Kopfschmerzen ein zentrales Merkmal der CPCS, werden aber bei der CTE nicht häufig berichtet. Auch wenn man darüber streiten kann, könnten Patienten der McKee-Stadien I und II Kopfschmerzen haben, was die Komplexität einer möglichen Überlappung von CTE & CPCS weiter erhöht (9). Die CPCS-Diagnose bleibt umstritten, da nicht klar ist, ob es sich um eine tauopathische Erkrankung handelt. Daher sind die Abgrenzungen zwischen CPCS und den klinischen Merkmalen der McKee-Stadien I und II nicht vollständig geklärt.
Eine eindeutige genetische Prädisposition für CTE ist nicht bekannt. Das ApoE4-Gen, der bekannteste Risikofaktor für die Alzheimer-Krankheit (25), wurde jedoch mit größeren kognitiven Defiziten und einer längeren Erholungsphase nach einer traumatischen Hirnverletzung (11) in Verbindung gebracht. In einer Studie an einer Gruppe von Boxern wurde über schwerere Folgen bei Personen berichtet, die mindestens ein ApoE4-Allel tragen (26). Umgekehrt könnte ApoE3 selbst bei Vorliegen einer fortschreitenden CTE-Pathologie eine neuroprotektive Wirkung haben (15). Ein weiterer vorgeschlagener Schutzfaktor, der mit einer günstigeren Erholung nach einer Schädel-Hirn-Trauma assoziiert ist, ist die kognitive Reserve, die anhand des prämorbiden IQ und des intrakraniellen Gesamtvolumens gemessen wird (27). Weitere genetische Kandidaten für weitere Untersuchungen sind das Gen für das Mikrotubuli assoziierte Protein Tau (MAPT), das Progranulin (GRN)-Gen und das Gen für den offenen Leserahmen von Chromosom 9 (C9ORF72) (11).
Der pathologische Synergismus von Tauopathie und Neuroinflammation wird zunehmend erkannt. Es wird angenommen, dass die extrazelluläre Sekretion von hyperphosphoryliertem Tau Mikroglia und Astrozyten aktiviert, was zur Produktion von entzündungsfördernden Zytokinen wie IL1ß und TNFa führt, die wiederum die Aktivierung von Tau-Kinasen wie p38 und cdk5 und eine weitere Tau-Phosphorylierung bewirken. Dieser Prozess führt zu einem ständigen Teufelskreis aus Tauopathie und Neuroinflammation (28). Angesichts des eindeutigen Zusammenhangs zwischen wiederholten traumatischen Hirnverletzungen und dem CTE-Risiko (1) könnte eine rechtzeitige Behandlung von Schädel-Hirn-Traumata die Entwicklung von CTE vermindern. Über die entzündungsfördernde Wirkung einer Schädel-Hirn-Trauma wurde bereits berichtet (13), und entzündungshemmende Wirkstoffe wie Minocyclin in Verbindung mit N-Acetylcystein, einem starken Antioxidans, die in akuten bis subakuten Zeitfenstern nach einer Schädel-Hirn-Trauma verabreicht werden, bieten ein vielversprechendes Therapieschema (29, 30). Die Entwicklung eines zeitsensitiven Protokolls, das dem Behandlungsalgorithmus für ischämische Schlaganfälle ähnelt, könnte das langfristige Ergebnis der Genesung nach einer Schädel-Hirn-Trauma messen und die Entwicklung einer späteren CTE-Pathologie verhindern (29).
Es gibt derzeit keine krankheitsmodifizierenden Medikamente für CTE, so dass die Prävention die wirksamste Methode zur Bekämpfung dieser schwächenden neurodegenerativen Erkrankung ist (31). Angesichts der Häufigkeit von Kopfkollisionen in Kontaktsportarten wie American Football erfordert die Prävention von Kopftraumata einen Kulturwandel in der Art und Weise, wie der Sport gelehrt und ausgeübt wird. Das Training sicherer Trainingstechniken, wie z. B. sicheres Tackling und Schlagen, und die Bestrafung rücksichtsloser Schläge werden messbare Vorteile bringen. Zu den weiteren Änderungen gehört die Schaffung eines sicheren Umfelds, in dem die Spieler ermutigt werden, den Schiedsrichtern, Trainern und Mannschaftsärzten Symptome zu melden. Darüber hinaus könnte die Erstellung eines neurokognitiven Ausgangsprofils als klinischer Referenzwert dienen, um Veränderungen im neuropsychiatrischen Erscheinungsbild der Spieler zu verfolgen. Es obliegt den Mannschaftsärzten, Spieler, die auch nur eine leichte, unkomplizierte Schädel-Hirn-Trauma erlitten haben, zur weiteren Untersuchung vom Spielfeld zu nehmen (32).
Es gibt eine Reihe von Herausforderungen im Zusammenhang mit CTE, die es zu bewältigen gilt. Obwohl die Inzidenz sportbedingter Gehirnerschütterungen Berichten zufolge zwischen 1,6 und 3,8 Millionen liegt, sind die Inzidenz und Prävalenz der CTE nach wie vor weitgehend unbekannt (33). Eine Erklärung für diese Unkenntnis ist vielleicht die Tatsache, dass Sportler, die kumulativen subkutiven Schlägen ausgesetzt sind, die eine ausreichende Kraft ausüben, um neuronale Schäden zu verursachen, aber zunächst keine offensichtlichen klinischen Symptome aufweisen, oft nicht rechtzeitig untersucht oder diagnostiziert werden (34). Groß angelegte prospektive Studien, wie z. B. die Verfolgung von Sportlern mit mehrfachen Schädel-Hirn-Traumata über einen bestimmten Zeitraum, würden unser Verständnis des natürlichen Verlaufs und der Phänomenologie der Krankheit erweitern. Die CTE rückt über die Massenmedien zunehmend in den Blickpunkt der Öffentlichkeit. Es bedarf kontinuierlicher Anstrengungen, um diese verheerende Krankheit zu diagnostizieren, zu bewerten und zu behandeln. Die exponentiellen Fortschritte bei den bildgebenden Verfahren und das Verständnis der neuropathologischen Mechanismen der Krankheit werden zu einer früheren Diagnose und rechtzeitigen Behandlungsmaßnahmen führen.
Beiträge des Autors
Der Autor bestätigt, dass er der einzige Autor dieser Arbeit ist und sie zur Veröffentlichung freigegeben hat.
Erklärung zu Interessenkonflikten
Der Autor erklärt, dass die Forschung in Abwesenheit jeglicher kommerzieller oder finanzieller Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Danksagungen
Der Autor dankt für die großartige Unterstützung, Anleitung und Mentorschaft von Dr. Stephen Strittmatter.
1. McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. (2009) 68:709-35. doi: 10.1097/NEN.0b013e3181a9d503
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. (2012) 4:134ra60. doi: 10.1126/scitranslmed.3003716
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Martland H. Dementia pugilistica. JAMA. (1928) 91:364-65. doi: 10.1001/jama.1928.02700150029009
CrossRef Volltext | Google Scholar
4. Millspaugh J. Dementia pugilistica. US Naval Med Bull. (1937) 35:303.
Google Scholar
5. Critchley M. Hommage a Clovis Vincent. Paris: Maloine (1949).
Google Scholar
6. Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, et al. Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA. (2017) 318:360-70. doi: 10.1001/jama.2017.8334
PubMed Abstract | CrossRef Full Text | Google Scholar
7. McKee AC, Stein TD, Kiernan PT, Alvarez VE. Die Neuropathologie der chronischen traumatischen Enzephalopathie. Brain Pathol. (2015) 25:350-64. doi: 10.1111/bpa.12248
PubMed Abstract | CrossRef Full Text | Google Scholar
8. McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. (2016) 131:75-86. doi: 10.1007/s00401-015-1515-z
PubMed Abstract | CrossRef Full Text | Google Scholar
9. McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. (2013) 136:43-64. doi: 10.1093/brain/aws307
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Jordan BD. Das klinische Spektrum von sportbedingten traumatischen Hirnverletzungen. Nat Rev Neurol. (2013) 9:222-30. doi: 10.1038/nrneurol.2013.33
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Stern RA, Daneshvar DH, Baugh CM, Seichepine DR, Montenigro PH, Riley DO, et al. Clinical presentation of chronic traumatic encephalopathy. Neurology. (2013) 81:1122-9. doi: 10.1212/WNL.0b013e3182a55f7f
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Gardner A, Iverson GL, McCrory P. Chronic traumatic encephalopathy in sport: a systematic review. Br J Sports Med. (2014) 48:84-90. doi: 10.1136/bjsports-2013-092646
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Montenigro PH, Baugh CM, Daneshvar DH, Mez J, Budson AE, Au R, et al. Clinical subtypes of chronic traumatic encephalopathy: literature review and proposed research diagnostic criteria for traumatic encephalopathy syndrome. Alzheim Res Ther. (2014) 6:68. doi: 10.1186/s13195-014-0068-z
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Mawdsley C, Ferguson F. Neurological disease in boxers. Lancet. (1963) 282:795-801. doi: 10.1016/S0140-6736(63)90498-7
CrossRef Full Text | Google Scholar
15. Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH. Chronische traumatische Enzephalopathie bei einem Spieler der National Football League. Neurosurgery. (2005) 57:128-34. doi: 10.1227/01.NEU.0000163407.92769.ED
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. (2011) 134:2456-77. doi: 10.1093/brain/awr179
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Omalu BI, Bailes J, Hammers JL, Fitzsimmons RP. Chronische traumatische Enzephalopathie, Suizide und Parasuizide bei amerikanischen Profisportlern: die Rolle des forensischen Pathologen. Am J Forensic Med Pathol. (2010) 31:130-2. doi: 10.1097/PAF.0b013e3181ca7f35
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Maroon JC, Winkelman R, Bost J, Amos A, Mathyssek C, Miele V. Chronische traumatische Enzephalopathie bei Kontaktsportarten: eine systematische Übersicht über alle gemeldeten pathologischen Fälle. PLoS ONE. (2015) 10:e0117338. doi: 10.1371/journal.pone.0117338
CrossRef Full Text | Google Scholar
19. Iverson GL. Chronische traumatische Enzephalopathie und Suizidrisiko bei ehemaligen Sportlern. Br J Sports Med. (2014) 48:162-4. doi: 10.1136/bjsports-2013-092935
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Iverson GL, Gardner AJ, McCrory P, Zafonte R, Castellani RJ.Iverson A critical review of chronic traumatic encephalopathy. Neurosci Biobehav Rev. (2015) 56:276-93. doi: 10.1016/j.neubiorev.2015.05.008
CrossRef Full Text | Google Scholar
21. Randolph C. Chronisch traumatische Enzephalopathie ist keine echte Krankheit. Arch Clin Neuropsychol. (2018) 33:644-8. doi: 10.1093/arclin/acy063
CrossRef Full Text | Google Scholar
22. Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R, et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature. (2019) 568:420-3. doi: 10.1038/s41586-019-1026-5
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Weir DR, Jackson JS, Sonnega A, et al. National Football League Player Care Foundation Study of Retired NFL Players. Ann Arbor: University of Michigan Institute for Social Research (2009).
Google Scholar
24. King NS, Kirwilliam S. Permanent post-concussion symptoms after mild head injury. Brain Inj. (2011) 25:462-70. doi: 10.3109/02699052.2011.558042
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Ward A, Crean S, Mercaldi CJ, Collins JM, Boyd D, Cook MN, et al. Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer’s disease: a systematic review and meta-analysis. Neuroepidemiology. (2012) 38:1-17. doi: 10.1159/000334607
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Jordan BD, Relkin NR, Ravdin LD, Jacobs AR, Bennett A, Gandy S. Apolipoprotein Eε4 associated with chronic traumatic brain injury in boxing. (1997) 278:136-40. doi: 10.1001/jama.278.2.136
CrossRef Full Text | Google Scholar
27. Stern Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. (2012) 11:1006-12. doi: 10.1016/S1474-4422(12)70191-6
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Laurent CL, Buée L, Blum D. Tau und Neuroinflammation: Welche Auswirkungen auf die Alzheimer-Krankheit und Tauopathien? Biomed J. (2018) 41:21-33. doi: 10.1016/j.bj.2018.01.003
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Sangobowale MA, Grin’kina NM, Whitney K, Nikulina E, St., Laurent-Ariot K, Ho JS, et al. Minocyclin plus N-Acetylcystein reduzieren Verhaltensdefizite und verbessern die Histologie mit einem klinisch sinnvollen Zeitfenster. J Neurotrauma. (2018) 35:907-17. doi: 10.1089/neu.2017.5348
CrossRef Full Text | Google Scholar
30. Haber M, James J, Kim J, Sangobowale M, Irizarry R, Ho J, et al. Minocyclin plus N-Acteylcystein induziert Remyelinisierung, schützt synergistisch Oligodendrozyten und modifiziert Neuroinflammation in einem Rattenmodell einer leichten traumatischen Hirnverletzung. J Cereb Blood Flow Metab. (2018) 38:1312-6. doi: 10.1177/0271678X17718106
PubMed Abstract | CrossRef Full Text | Google Scholar
31. Livingston G, Sommerlad A, Orgeta V, Costafreda SG, Huntley J, Ames D, et al. Dementia prevention, intervention and care. Lancet. (2017) 390: 2673-734. doi: 10.1016/S0140-6736(17)31363-6
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Saulle M, Greenwald BD. Chronische traumatische Enzephalopathie: ein Überblick. Rehabil Res Pract. (2012). 2012: 816069. doi: 10.1155/2012/816069
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Langlois JA, Rutland-Brown W, Wald MM. Epidemiologie und Auswirkungen von traumatischen Hirnverletzungen. J Head Trauma Rehabil. (2006) 21:375-8. doi: 10.1097/00001199-200609000-00001
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Martini D, Eckner J, Kutcher J, Broglio SP. Subconcussive head impact biomechanics: comparing different offensive schemes. Med Sci Sports Exerc. (2013) 45:755-61. doi: 10.1249/MSS.0b013e3182798758
PubMed Abstract | CrossRef Full Text | Google Scholar