Key Words: progressive hemifacial atrophy, Parry-Romberg syndrome
Progressive hemifacial atrophy-or Parry-Romberg syndrome-is a rare disease, classified as one of the forms of localized morphea or scleroderma. La sua causa è sconosciuta. È caratterizzata da atrofia della pelle, del grasso, dei muscoli e delle strutture osteocartilaginee sottostanti, che di solito colpisce unilateralmente il viso e il collo, ed è associata a sintomi neurologici (epilessia secondaria) e coinvolgimento di altri organi e sistemi. Il suo decorso è lento e progressivo e inizia nelle prime due decadi di vita. È stata osservata una predilezione per il sesso femminile. Riportiamo il caso di una bambina di 10 anni diagnosticata all’ospedale Hipólito Unánue di Tacna, in Perù. La conoscenza di questa condizione è importante nella diagnosi differenziale delle morfee localizzate o della sclerodermia.
Idee chiave
- L’atrofia emifacciale progressiva è una malattia rara, con un’incidenza di tre casi su 100 000 persone all’anno; è più frequente nelle donne e la sua eziologia è sconosciuta.
- È caratterizzata dalla progressione dell’atrofia, di solito unilaterale sul lato sinistro del viso, che compromette le strutture osteocartilaginee.
- Nel caso riportato, il paziente ha presentato la lesione sul lato destro, un reperto poco frequente.
- La conoscenza di questa malattia è importante per la diagnosi differenziale di morfea o sclerodermia localizzata.
Introduzione
L’atrofia emifacciale progressiva, nota anche come sindrome Parry-Romberg, è stata descritta per la prima volta da Caleb Hillier Parry e Moritiz Heinrich Romberg nel 1825 e 1846, ed è caratterizzata da una progressiva atrofia della pelle e dei tessuti sottostanti, che colpisce il viso e il collo solitamente unilateralmente,,. La sua incidenza è di circa tre casi su 100.000 persone all’anno. Anche se questa rara malattia colpisce normalmente solo il viso e il collo unilateralmente, occasionalmente si manifesta bilateralmente e colpisce anche il tronco, le braccia e le gambe. Il suo decorso è progressivo, lento e insorge principalmente tra i due e i 20 anni,.
La patogenesi di questa malattia è sconosciuta,, tuttavia sono state proposte diverse teorie, tra cui cause immunologiche, traumatiche, infettive, endocrinologiche, neurologiche e genetiche. L’epilessia è l’anomalia più frequente associata alla sindrome di Perry-Romberg.
Segnaliamo una bambina di 10 anni con diagnosi di atrofia emifacciale progressiva presso l’ospedale Hipolito Unanue di Tacna, Perù, nel 2015. Per quanto ne sappiamo, questo è il terzo caso riportato in Perù e il primo caso a Tacna.
Relazione del caso
Questo è il caso di una paziente di 10 anni di Tacna (una città del Perù meridionale al confine con la Bolivia e il Cile). È il prodotto di una seconda gestazione ed è nata a termine con parto cesareo. I suoi genitori non sono consanguinei e i suoi parenti stretti sono presumibilmente sani. Nel periodo perinatale non è stata rilevata alcuna malattia rilevante. Lo sviluppo fisico e motorio era adeguato.
La nostra paziente è arrivata al pronto soccorso dell’ospedale Hipólito Unánue presentando perdita di coscienza e crisi tonico-cloniche che si sono evolute in stato epilettico focale. Per questo motivo, la ragazza è stata ricoverata per ulteriori studi e trattamenti.
All’esame fisico, abbiamo osservato la presenza di due lesioni atrofiche nel viso: la prima e più lunga lesione, situata sulla linea mediale del viso, era di otto centimetri di lunghezza, due centimetri di larghezza e tra uno e due millimetri di profondità, diffondendosi dal trichion all’ala destra del naso; una lieve iperpigmentazione è stata notata al suo apice (Figure 1 e 2, indicata dalla freccia rossa). La seconda lesione atrofica, situata lateralmente alla prima, si estendeva dall’attaccatura dei capelli frontale destra all’arco sopracciliare destro, dove si osservava una zona di madarosi, ed era lunga quattro centimetri, larga un centimetro e profonda sette millimetri (figure 1 e 2, lesione indicata dalla freccia verde).
Figura 1. Lesioni atrofiche sul viso.
Figura 2. Zona di madarosi.
Nella regione parietale destra, è stata trovata una zona di alopecia (Figura 3). Allo stesso tempo, abbiamo osservato anisocoria e midriasi della pupilla dell’occhio destro, ma era ancora fotoreattiva. Sono stati notati anche una moderata ptosi palpebrale destra, un’ulcera corneale destra, una dislocazione del cristallino destro e un’ametropia. Nessuna anomalia è stata trovata nella retina. L’atrofia dell’ala destra del naso è stata notata, insieme ad un setto deviato, una commissura orale destra elevata, un’accentuazione del solco nasogenieno destro (Figura 2, freccia gialla), e una posizione anomala dei denti (Figura 4). Il resto dell’esame era normale.
Figura 3. Zona alopecica nella regione parietale destra.
Figura 4. Posizione anomala dei denti.
All’età di tre anni, il nostro paziente ha subito un trauma cerebrale con perdita di coscienza e convulsioni. A quel tempo, questi sintomi sono stati ritenuti causati dal trauma. Un anno dopo, i suoi genitori hanno notato la presenza e la progressione dell’anomalia del viso (descritta nei paragrafi precedenti); tuttavia, non hanno cercato l’assistenza medica perché hanno pensato che fosse una conseguenza del trauma precedente.
Gli esami di laboratorio che abbiamo eseguito per la paziente hanno mostrato una lieve leucocitosi con neutrofilia (18,3 x 103 cellule per mm3, 79% neutrofili) e glicemia elevata (149,8mg/dl). Questi risultati sono stati spiegati da convulsioni. Abbiamo ottenuto risultati negativi (normali) per i test degli anticorpi anti-nucleo, degli anticorpi anti-citoplasma dei neutrofili, dell’antigene di superficie dell’epatite B e delle IgM contro il virus dell’epatite A.
La tomografia computerizzata (CT) del cervello ha mostrato calcificazioni ed edema circostante nei gangli della base destra: nel putamen e nella testa del nucleo caudato (Figura 5). Nella ricostruzione CT 3D del cranio, abbiamo trovato asimmetria delle ossa facciali: atrofia dell’osso mascellare destro e dell’osso frontale, posizionamento anomalo dei denti, deviazione del setto nasale e crescita eccessiva del turbinato inferiore sinistro (Figura 6).
Figura 5. Calcificazioni nei gangli della base del cervello.
Figura 6. Deviazione del setto nasale con convessità sinistra.
Una volta ottenuti i dati dell’anamnesi, dell’esame fisico e degli esami diagnostici, l’autore principale di questo articolo ha effettuato una ricerca esaustiva nei database delle malattie rare (come Mendelian Inheritance Online in Man – OMIM, disponibile su www.omim.org), concludendo che la condizione della ragazza era compatibile con la sindrome di Parry-Romberg o l’atrofia emifacciale progressiva.
Avendo raggiunto un adeguato controllo delle crisi, la paziente è stata dimessa dall’ospedale; tuttavia, è sotto osservazione medica da un neurologo, pediatra, oculista e psicologo. Questo team multidisciplinare ha lo scopo di migliorare la qualità della vita della nostra paziente.
Discussione
La sindrome di Parry-Romberg è una malattia rara caratterizzata da una progressiva atrofia della pelle, dei muscoli e delle ossa che compromette il viso, di solito unilateralmente, e in alcuni casi è accompagnata da anomalie della dentizione, lingua e palato. Anche se la sindrome di Parry-Romberg è di solito unilaterale, è possibile trovare pazienti con danni bilaterali, trovando anche quelli con tronco, braccia e gambe compromesse.
L’incidenza di questa malattia è di tre casi su 100 000 pazienti all’anno ed è più comune nelle donne con proporzione di 3:1. Si presenta tipicamente nei primi venti anni di vita.
Questa malattia è solitamente classificata come esclerodermia localizzata lineare o morfea. Nella letteratura mondiale, abbiamo trovato riferimenti alla sindrome di Parry-Romberg come morfea in coup de sabre (“colpo di sciabola”, a causa della somiglianza dell’atrofia della pelle alla ferita causata dalla sciabola). Tuttavia, dalla nostra comprensione, queste condizioni sono due entità diverse, in quanto i pazienti che soffrono della sindrome Parry-Romberg non mostrano caratteristiche istologiche di sclerosi dermica, mentre i pazienti con morfea in colpo di sciabola la mostrano,.
L’eziologia della sindrome Parry-Romberg è ancora sconosciuta; tuttavia, l’ipotesi più accettata è l’eziologia autoimmune,. Le vie infiammatorie Th1 e Th17 sono di grande importanza nella fisiopatologia di questa malattia, almeno nelle prime fasi. La via infiammatoria Th2 è importante nella fase tardiva, la fase fibrotica. Questa risposta infiammatoria dei tessuti potrebbe essere iniziata da un evento traumatico accidentale, un intervento chirurgico o anche un trauma ostetrico. La teoria neurologica suggerisce che questa malattia è il risultato di una migrazione disorganizzata delle cellule della cresta neurale che compromette uno o più rami del nervo trigemino.
Alcuni rapporti affermano che la sindrome Parry-Romberg è una malattia genetica autosomica dominante. Inoltre, è stato detto che l’atrofia emifacciale progressiva è legata alla malattia di Lyme, Borrelia burgdorferi, e ad altre infezioni come la sifilide, la rosolia, la tubercolosi e il virus dell’epatite B.
In questo caso, il paziente ha sviluppato la malattia dopo una caduta, acquisendo un trauma cranico. Non c’erano precedenti di malattia nella famiglia. I suoi test di laboratorio per la presenza di anticorpi antinucleari e di epatite virale erano negativi. Non abbiamo valutato la nostra paziente per la malattia di Lyme perché il Perù non è una zona endemica per questa malattia, e lei ha negato di aver viaggiato in regioni endemiche.
La sindrome di Parry-Romberg coinvolge dermatomi di uno o più rami del nervo trigemino e di solito compromette la metà sinistra del viso,. Può presentare un certo grado di aumento della pigmentazione e della luminosità della pelle. L’alopecia può essere vista in qualsiasi area del cuoio capelluto ma di solito nella regione frontoparietale,.
M. Wong, nella sua serie di casi, dimostra una predilezione della malattia per la metà sinistra del viso. Carlos Galarza nei suoi due casi riportati da Cuzco, Perù, ha anche dimostrato l’atrofia della pelle sul lato sinistro. Nel frattempo, il nostro paziente ha presentato un’atrofia cutanea localizzata nella metà destra del viso, che si estendeva dalle regioni frontali a quelle mandibolari, accompagnata da lieve iperpigmentazione, deviazione del setto, distribuzione anomala dei denti e localizzazione parietale dell’alopecia.
Le manifestazioni oculari della sindrome di Parry-Romberg possono essere presenti prima, durante o dopo la comparsa dell’atrofia cutanea e possono includere: enoftalmo con conseguente diplopia (causata dall’atrofia del grasso nello spazio retrobulbare), occhio secco o uveite. Ci sono state anche segnalazioni di pigmentazione palpebrale, fotofobia, strabismo, atrofia dell’iride, panuveite, corpo ciliare ipotonico, vasculite retinica, edema e distacco della retina, ecc.
La valutazione neurologica può mostrare una paralisi del nervo oculomotore che si manifesta con anomalie pupillari, anisocoria, midriasi o miosi sul lato colpito e coinvolgimento del nervo ottico che può portare a neuroretinite e papillite. La nostra paziente, fin dalla sua prima visita al dipartimento di emergenza, ha sofferto di grave anisocoria e midriasi dell’occhio destro, un’anomalia della reazione pupillare, e con il progredire della malattia, sono apparse altre manifestazioni: dislocazione del cristallino destro, ulcera corneale, enoftalmo grave e ametropia.
Dalla Costa, nella sua revisione mondiale di 205 pazienti con diagnosi di sindrome di Parry-Romberg, ha riferito che il 50% dei pazienti aveva sintomi di un sistema nervoso centrale compromesso e il 15% aveva crisi epilettiche, mal di testa, dolore facciale, coinvolgimento dei nervi cranici ed emiplegia.
La manifestazione neurologica più frequente della sindrome di Parry-Romberg è l’epilessia (60,5% di frequenza). Le crisi epilettiche focali omolaterali alle calcificazioni cerebrali sono comuni (osservate nel 50% dei casi totali). Con una frequenza del 33%, l’epilessia secondaria è molto difficile da trattare. Possono essere presenti anche cefalea (44%) e nevralgia del trigemino (8,5%).
Le calcificazioni cerebrali intraparenchimali sullo stesso lato delle anomalie facciali sono i risultati più frequenti nella tomografia computerizzata e nella risonanza magnetica. Sono state descritte iperintensità della materia bianca, infrazione focale del corpo calloso, emiatrofia cerebrale e miglioramento leptomeningeo, aneurismi intracranici e malformazioni vascolari e microemorragie cerebrali. Non c’è correlazione tra il neuroimaging e le caratteristiche facciali di questa malattia.
Anche se le caratteristiche neurologiche e oculari sono le manifestazioni più frequenti della sindrome di Parry-Romberg, è possibile trovare anomalie cardiache (cardiomiopatia ipertrofica), anomalie endocrine (ipertiroidismo, ipotiroidismo), malattie autoimmuni (cirrosi biliare primaria, artrite reumatoide, sclerosi multipla) e malattie congenite, come la sindrome di Poland, microftalmie e malformazioni renali.
L’epilessia secondaria e il mal di testa erano le caratteristiche neurologiche predominanti nel nostro caso. Le crisi epilettiche erano state ben controllate. I risultati della tomografia e della risonanza sono stati descritti come calcificazioni nei gangli della base: putamen e nucleo caudato. Non abbiamo trovato altre anomalie oltre a quelle descritte sopra.
L’obiettivo del trattamento è quello di fermare la fase attiva della malattia. Questo può essere raggiunto con la prescrizione di metotrexato a 0,3 – 1mg/kg/settimana per uno o due anni, e può essere associato al prednisone durante i primi tre mesi considerando l’effetto ritardato del metotrexato sull’infiammazione e la fibrosi.
La sindrome di Parry-Romberg ha un grande impatto biopsicosociale sulla vita del paziente a causa delle limitazioni funzionali ed estetiche. Sono stati proposti alcuni approcci chirurgici per ripristinare la simmetria facciale. I casi lievi e moderati possono essere trattati con filler di silicone, collagene, impianti di polietilene poroso e innesto di grasso autologo. L’innesto di cartilagine, ossa, grasso e pelle sono opzioni di trattamento per i casi più gravi. Lo scopo del trattamento psicologico è la reintegrazione sociale dei pazienti.
La prognosi della malattia dipende dall’età del paziente, essendo più grave quando i cambiamenti atrofici iniziano in età adulta (dai 20 anni in su). La gravità dell’atrofia, il danno cerebrale e la scarsa risposta al trattamento sono maggiori nei pazienti di età superiore ai 20 anni. Questi fattori determinanti ci danno l’approccio globale alle sequele e al trattamento che deve essere considerato per ottenere migliori risultati terapeutici.
Conclusioni
L’emi atrofia facciale progressiva, o sindrome di Parry-Romberg, è una malattia rara e progressiva di causa sconosciuta che colpisce soprattutto le donne. Ci sono pochi casi riportati di questa sindrome in Perù, e questo è il primo caso nella regione di Tacna.
È caratterizzata dalla progressione dell’atrofia, di solito unilaterale, essendo più frequente sul lato sinistro del viso e coinvolgendo strutture osteocartilaginee. Nel nostro caso, l’anomalia cutanea è stata trovata sul lato destro; questo è un reperto poco frequente.
La conoscenza di questa malattia diventa rilevante nella diagnosi differenziale delle lesioni atrofiche in un lato del viso. Tra le malattie con le quali deve essere fatta la diagnosi differenziale, spiccano: la malattia del colpo di sciabola, il lupus profondo, l’atrofia da steroidi e altri tipi di morfea.
Contributi degli autori
EOL: autore principale, diagnosi e follow-up del paziente, scrittura e revisione critica dell’articolo, approvazione finale dell’articolo e assunzione di responsabilità del manoscritto. SDA: autore, scrittura e importanti contributi all’articolo e assunzione di responsabilità per il manoscritto. PDA: coautore, contributi importanti all’articolo
Conflitto di interessi
Nessuno degli autori dichiara di avere conflitti di interesse con l’argomento di questo articolo.
Fonte di finanziamento
Il finanziamento nell’elaborazione dell’articolo è proprio.
Aspetti etici
Gli autori hanno ottenuto il consenso informato dei genitori della paziente (essendo questa minorenne) per la pubblicazione di questo articolo e delle immagini che lo accompagnano.
Dati
Dichiariamo la disponibilità per la consegna dei dati su richiesta.
Dagli editori
La versione originale di questo manoscritto è stata presentata in spagnolo. Questa versione inglese è stata presentata dagli autori ed è stata leggermente copiata dal Journal.

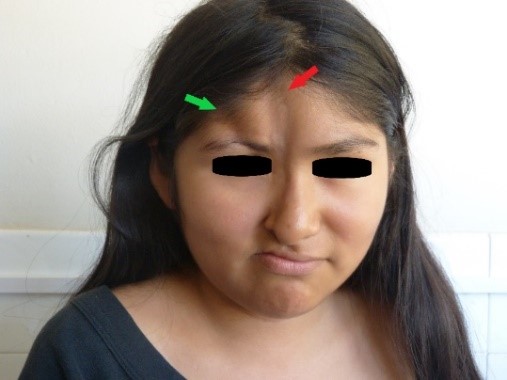 Figura 1. Lesioni atrofiche sul viso.
Figura 1. Lesioni atrofiche sul viso. 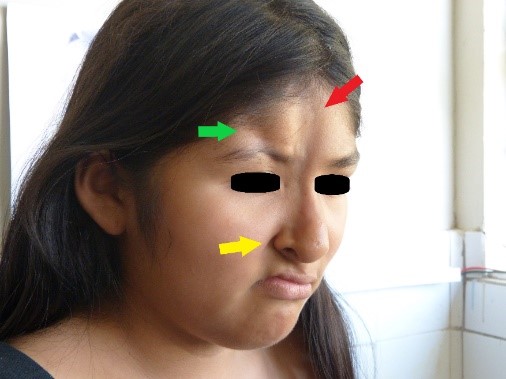 Figura 2. Zona di madarosi.
Figura 2. Zona di madarosi. 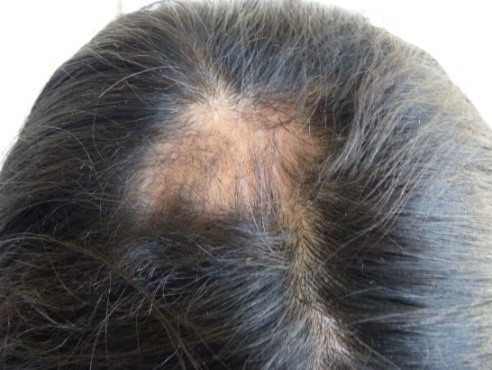 Figura 3. Zona alopecica nella regione parietale destra.
Figura 3. Zona alopecica nella regione parietale destra. 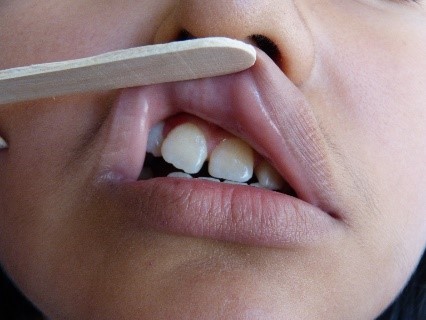 Figura 4. Posizione anomala dei denti.
Figura 4. Posizione anomala dei denti. 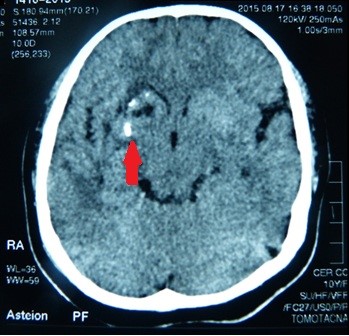 Figura 5. Calcificazioni nei gangli della base del cervello.
Figura 5. Calcificazioni nei gangli della base del cervello. 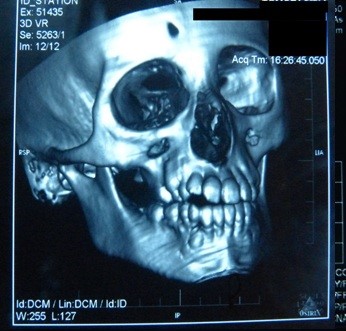 Figura 6. Deviazione del setto nasale con convessità sinistra.
Figura 6. Deviazione del setto nasale con convessità sinistra.