L’encefalopatia traumatica cronica (CTE) è una malattia neurodegenerativa associata alla proteina tau. C’è stato un aumento delle diagnosi di CTE negli atleti, specialmente nei giocatori di football americano, così come nei veterani militari in combattimento (1, 2). Anche se la CTE è stata riconosciuta pubblicamente in tempi relativamente recenti, è stata descritta per la prima volta come sindrome “punch drunk” in un classico articolo di Martland et al. (3). La relazione era incentrata su un certo numero di pugili che avevano subito ripetuti colpi alla testa nel corso della loro carriera, e si presentavano sia con sintomi psichiatrici che con gravi deficit di memoria e neurocognitivi, analoghi ai tipici pazienti con demenza (3). La nomenclatura della malattia si è evoluta in “demenza pugilistica” (4), e infine CTE nel 1949 (5).
CTE ha una caratteristica neuropatologica unica, composta da accumulo di tau fosforilato (p-tau) in sulci e regioni peri-vascolari, microgliosi e astrocitosi. Questi cambiamenti patologici portano a una progressiva neurodegenerazione debilitante. Sulla base del modello di progressione patologica, il CTE è diviso in quattro stadi rispettivi (Figura 1). Nello stadio I del CTE, il cervello appare grossolanamente normale, ma la p-tau si trova in un numero finito di punti, spesso nelle cortecce laterali e frontali, così come prossimale ai piccoli vasi sanguigni nella profondità dei solchi. Ci potrebbe essere un numero esiguo di grovigli neurofibrillari (NFT) e neuriti nel locus coeruleus. Nello stadio II, si possono notare anomalie macroscopiche localizzate. Su sezioni anatomiche grossolane e neuroimaging, si osserva l’allargamento dei ventricoli laterali, il cavum septum pellucidum con o senza fenestrazione, così come il pallore del locus coeruleus e della substantia nigra. Ci sono focolai multipli di p-tau all’interno della profondità dei solchi, e c’è un modello di diffusione emergente. Nello stadio III, la maggior parte delle sezioni patologiche grossolane mostrano anomalie macroscopiche. C’è una perdita di peso globale del cervello, una lieve atrofia del lobo frontale e del lobo temporale, e una dilatazione dei ventricoli. Una metà dei pazienti con CTE mostra anomalie del setto, incluso il cavum septum pellucidum. La patologia P-tau si diffonde, coinvolgendo le cortecce frontale, temporale, parietale e insulare. Nello stadio IV, la riduzione del peso del cervello è drammatica, e sono stati riportati pesi del cervello di 1.000 g (rispetto ai 1.300-1.400 g dei cervelli normali). C’è una profonda atrofia dei lobi frontali e temporali mediali, così come dei talami anteriori. C’è anche atrofia dei tratti di materia bianca. La maggior parte dei pazienti al quarto stadio ha anomalie del setto. La diffusione della p-tau colpisce la maggior parte delle regioni, compresa la corteccia calcarina (7, 8). Anomalie nella proteina fosforilata 43 kDa TAR legante il DNA (TDP-43) è anche visto nella maggior parte dei pazienti CTE. La patologia parenchimale TDP-43 è anche di natura progressiva simile al modello anatomico di diffusione della p-tau. L’immunoreattività TDP-43 si trova in quasi tutti i casi di malattia di stadio IV (7).

Figura 1. Le immagini qui sopra sono la rappresentazione dei quattro stadi di McKee del CTE.
Il fenotipo clinico del CTE deve ancora essere chiaramente definito. I paragrafi seguenti delineano i tentativi di caratterizzazione dei sintomi CTE nelle varie fasi del processo della malattia (Tabella 1). Secondo la classificazione di McKee, nello stadio I, un tipico paziente con CTE è asintomatico, o può lamentare lievi deficit di memoria a breve termine e sintomi depressivi. Può essere osservata una lieve aggressività. Nello stadio II, l’umore e i sintomi comportamentali potrebbero includere scoppi comportamentali e sintomi depressivi più gravi. Nella fase III, i pazienti presentano tipicamente più deficit cognitivi, tra cui perdita di memoria, deficit del funzionamento esecutivo, disfunzione visuospaziale e apatia. Nello stadio IV, i pazienti presentano deficit linguistici avanzati, sintomi psicotici tra cui paranoia, deficit motori e parkinsonismo.
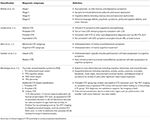
Tabella 1. Classificazioni cliniche proposte per il CTE.
Jordan et al. (10) furono tra i primi a caratterizzare clinicamente la malattia. Hanno diviso le presentazioni cliniche della CTE in tre domini: comportamentale/psichiatrico, cognitivo e motorio. Il dominio comportamentale e psichiatrico comprendeva l’aggressività, la depressione, l’apatia, l’impulsività, i deliri, compresa la paranoia, e la suicidalità. Il dominio cognitivo comprendeva una diminuzione dell’attenzione e della concentrazione, deficit di memoria, deficit del funzionamento esecutivo, disfunzioni visuospaziali, deficit del linguaggio e demenza. Infine, le caratteristiche motorie consistevano in disartria, anomalie dell’andatura, atassia e incoordinazione, spasticità e caratteristiche del parkinsonismo come i tremori. Sulla base di queste caratteristiche cliniche, così come le informazioni neuropatologiche esistenti, sono stati definiti quattro sottotipi diagnostici, vale a dire “Definito,” Probabile,” “Possibile,” e “Improbabile” CTE.
Stern et al. (11), e relativi rapporti di casi (14, 15), differivano nella loro descrizione di un tipico paziente CTE, concettualizzando la presentazione clinica in due sottotipi distinti. Il primo sottotipo mostrava principalmente cambiamenti comportamentali e dell’umore, mentre l’altro presentava principalmente un deterioramento cognitivo. La grande maggioranza del sottotipo umore/comportamento ha sviluppato deficit cognitivi con il progredire della malattia. Tuttavia, relativamente pochi pazienti del gruppo cognitivo hanno mostrato alterazioni dell’umore o del comportamento durante il corso della loro malattia. Nello studio di Stern et al. (11), i pazienti del gruppo cognitivo avevano una probabilità significativamente maggiore di sviluppare la demenza. Erano anche significativamente più anziani al momento della diagnosi rispetto ai pazienti del gruppo umore/comportamento. Il sottogruppo comportamentale dei pazienti con CTE può assomigliare ai pazienti affetti da demenza frontotemporale con variante comportamentale (bvFTD), il che rende la diagnosi clinica più difficile. Tuttavia, le manifestazioni comportamentali tipiche della bvFTD, come l’apatia e la disinibizione, spesso non si vedono nei pazienti con CTE (11, 16). Data l’eterogeneità intrinseca della bvFTD, così come la natura tauopatica simile di entrambe le malattie, distinguere la bvFTD e la CTE pone una sfida diagnostica.
Tra i sintomi comportamentali della CTE, l’associazione tra suicidio e CTE rimane un argomento sotto esame in letteratura. Studi precedenti, come la serie di cinque atleti professionisti con una diagnosi confermata di CTE riportata da Omalu et al. (17), avevano suggerito una forte relazione tra CTE e suicidio. Gli autori hanno inoltre suggerito che l’eziologia del comportamento suicida/parasuicida nella popolazione CTE potrebbe essere in parte dovuta alla tauopatia sotto forma di grovigli neurofibrillari e fili neuritici nei nuclei strategici del cervello limbico come il locus ceruleus. Maroon et al. (18), ha esaminato 153 casi patologicamente confermati di CTE pubblicati tra 1954 e 2013. Hanno riportato la prevalenza di suicidio nella popolazione CTE e morti accidentali per essere 11.7 e 17.5%, significativamente superiore ai livelli della popolazione generale di 1.5 e 4.8%, rispettivamente (18). I sostenitori del punto di vista opposto suggeriscono che i suicidi sono stati segnalati soprattutto nelle fasi iniziali della CTE, e l’associazione tra la progressione della malattia e il suicidio rimane poco chiara in questo momento (19).
In una meta-analisi di 158 studi di casi di Gardner et al. (12), i sintomi clinici della CTE sono stati suddivisi in “classici” vs. sintomi “moderni” della CTE, per fare una distinzione tra una vecchia descrizione dei casi di CTE incentrata soprattutto sui pugili rispetto a una descrizione clinica più evoluta che si applica anche ai giocatori professionisti di football americano. Mentre i sintomi “classici” della CTE includevano tipicamente disartria, difficoltà di movimento e più tardi progressione verso deficit di memoria, il quadro “moderno” della CTE includeva anche sintomi neuropsichiatrici, come sintomi depressivi, paranoia, ritiro sociale e isolamento, compromissione del giudizio e aggressività. I deficit cognitivi come il declino della memoria, la disfunzione esecutiva, il linguaggio e i deficit di elaborazione delle informazioni emergono più tardi nel corso del processo di malattia (12).
Siccome la definizione di CTE dipende principalmente dalle caratteristiche patologiche, esiste un termine clinico alternativo proposto da Montenigro et al. (13) di sindrome da encefalopatia traumatica (TES), che descrive le conseguenze cliniche di TBI ripetitive. Gli autori hanno basato questa classificazione su una revisione di 202 casi pubblicati. TES è una diagnosi più comprensiva e può essere suddivisa in quattro sottocategorie, tra cui TES variante comportamentale/umorale, TES variante cognitiva, TES variante mista e TES demenza. La diagnosi TES proposta si basa sull’esistenza di cinque criteri generali, tre caratteristiche cliniche fondamentali e nove caratteristiche di supporto. Utilizzando i biomarcatori esistenti* (Tabella 1), sono stati proposti ulteriori qualificatori diagnostici, che includevano “Probabile”, “Possibile” e “Improbabile” CTE (9, 13). La diagnosi TES proposta conteneva anche qualificatori temporali e includeva “decorso progressivo”, “decorso stabile” e “decorso sconosciuto/inconsistente”. Se la presentazione clinica includeva anche segni motori come il parkinsonismo, veniva aggiunto anche il modificatore “con caratteristiche motorie”.
Come cresce la nostra comprensione della CTE, ci sono una serie di sfide e critiche che devono essere affrontate. Un’ipotesi alternativa al fenomeno CTE è la teoria della “riserva cognitiva” diminuita. La teoria afferma che il neurotrauma ripetitivo porta a una riduzione della riserva cognitiva e all’accelerazione dello sviluppo di un disturbo neurodegenerativo sottostante (20, 21). Se questa teoria fosse vera, implicherebbe che il CTE e l’AD sono sullo stesso spettro neuropatologico. Questa affermazione merita ulteriori analisi. Come nell’AD, anche nella CTE le isoforme di Tau consistono in un mix di isoforme a tre ripetizioni (3R) e a quattro ripetizioni (4R). Tuttavia, secondo un recente rapporto di Falcon et al. (22), i filamenti di tau estratti dal cervello di pazienti con CTE contengono anche un’unica regione a ß-elica con una cavità idrofoba, che non è presente nel cervello dei pazienti con AD. La cavità contiene un ulteriore cofattore che si pensa abbia un ruolo funzionale nella propagazione della tau. Falcon et al. (22) suggeriscono che la posizione delle inclusioni di tau in prossimità dei vasi sanguigni, suggerisce che i cofattori necessari per l’assemblaggio di tau possono attraversare la barriera emato-encefalica dopo un trauma cranico. Gli autori sostengono inoltre che il fatto che il trauma cerebrale porta a CTE solo in un sottogruppo di popolazione ferita, potrebbe essere correlato al livello più elevato di cofattori negli individui più suscettibili. Questi cofattori potrebbero fornire un bersaglio terapeutico per la prevenzione dell’assemblaggio di tau e lo sviluppo di CTE post trauma (22).
Una teoria alternativa propone che i sintomi psichiatrici come la depressione e la rabbia riportati nei pazienti CTE sono indipendenti dal processo di malattia CTE e sono riportati in modo cofondato. I sostenitori di questa ipotesi hanno citato studi precedenti come quello riportato da Weir et al. (23), in cui è stato chiesto a 1.063 ex giocatori della NFL se avessero avuto attacchi di rabbia. È stato riportato che il 30,7% dei giocatori di età compresa tra i 30 e i 49 anni e il 29,3% dei giocatori di età pari o superiore ai 50 anni hanno riportato attacchi di rabbia. Tuttavia, gli autori hanno anche notato che le misure di rabbia riportate erano effettivamente inferiori a quelle riportate per la popolazione generale degli Stati Uniti, che era del 54,8% per gli uomini tra i 30 e i 49 anni, e del 47,2% per gli uomini sopra i 50 anni (23). Anche se gli argomenti relativi alla comorbilità dei sintomi psichiatrici e delle malattie neurodegenerative come la CTE, sono difficili da verificare sulla base dei risultati di neuroimaging e neuropatologici, si possono applicare argomenti simili ai sintomi psichiatrici di qualsiasi condizione neurodegenerativa come AD, bvFTD, Parkinson Disease (PD) o sclerosi laterale amiotrofica (ALS).
Un’altra importante fonte di confusione diagnostica è la delineazione clinica tra la CTE e la sindrome post-concussiva prolungata (PCS), soprattutto in considerazione dei rapporti precedenti che indicano che ~ 10-20% degli individui che subiscono commozioni cerebrali, sperimentano sintomi prolungati. La sindrome postconcussiva cronica (CPCS) si riferisce alla persistenza dei sintomi PCS che portano alla compromissione delle prestazioni funzionali e spesso atletiche che durano più di 1 anno. I sintomi della CPCS includono mal di testa, vertigini, deficit di attenzione, memoria e funzionamento esecutivo, depressione e sintomi di irritabilità (10). King e Kirkwilliam hanno coniato il termine “PCS permanente” per riferirsi a quelli con sintomi PCS che persistono in media 6,9 anni dopo la commozione cerebrale iniziale. Inoltre, hanno riferito che un numero significativo di pazienti con PCS permanente (40-59%) aveva anche condizioni neuropsichiatriche premorbose o postmorbose come depressione, ansia, PTSD, e/o dolore (24). Come sostenuto da Jordan et al. (10), la PCS è clinicamente distinguibile dalla CTE, sulla base della sua relazione temporale all’evento concussivo acuto. Un’anamnesi temporale accurata e approfondita rimane fondamentale nella valutazione neurologica. Inoltre, la cefalea è una caratteristica centrale della CPCS ma non è comunemente riportata nella CTE. Anche se discutibile, i pazienti degli stadi I e II di McKee potrebbero presentare mal di testa, aggiungendo ulteriormente la complessità della possibile sovrapposizione di CTE & CPCS (9). La diagnosi di CPCS rimane controversa, poiché non è chiaro se sia di natura tauopatica. Quindi, le linee di demarcazione della CPCS e le caratteristiche cliniche degli stadi I e II di McKee non sono completamente consolidate.
Predisposizioni genetiche chiare alla CTE non sono state riportate. Tuttavia, il gene ApoE4, il fattore di rischio più noto per il morbo di Alzheimer (25), è stato associato a maggiori deficit cognitivi e un periodo di recupero più prolungato dopo una lesione cerebrale traumatica (TBI) (11). Uno studio su un gruppo di pugili ha riportato esiti più gravi negli individui che portano almeno un allele ApoE4 (26). Al contrario, ApoE3 potrebbe conferire neuroprotezione, anche in presenza di una patologia progressiva CTE (15). Un altro fattore protettivo proposto associato con più favorevole recupero post TBI è la riserva cognitiva, come misurato dal QI premorboso e il volume intracranico totale (27). Altri candidati genetici per ulteriori studi includono il gene della proteina tau associata al microtubulo (MAPT), il gene della progranulina (GRN) e il gene del cromosoma nove open reading frame 72 (C9ORF72) (11).
La sinergia patologica di tauopatia e neuroinfiammazione è sempre più riconosciuta. Secrezione extracellulare di tau iperfosforilato è pensato per attivare microglia e astrociti, portando alla produzione di citochine pro-infiammatorie come IL1ß, e TNFa, a sua volta portando all’attivazione di tau chinasi come p38 e cdk5, e ulteriore fosforilazione tau. Questo processo crea un vizioso ciclo perpetuo di tauopatia e neuroinfiammazione (28). Data la robusta associazione tra lesioni cerebrali traumatiche ripetitive e rischio di CTE (1), il trattamento tempestivo di TBI potrebbe diminuire lo sviluppo di CTE. La natura pro-infiammatoria della TBI è stata precedentemente riportata (13), e gli agenti anti-infiammatori come la minociclina con N-acetilcisteina, un potente anti-ossidante, somministrato in acuto a finestre temporali subacute post TBI, offrono un regime terapeutico promettente (29, 30). Lo sviluppo di un protocollo sensibile al tempo, simile all’algoritmo di trattamento per l’ictus ischemico, potrebbe potenzialmente misurare il risultato a lungo termine nel recupero post TBI e la prevenzione dello sviluppo della successiva patologia CTE (29).
Non ci sono attualmente farmaci che modificano la malattia per CTE, rendendo la prevenzione il modo più efficace di combattere questa malattia neurodegenerativa debilitante (31). Data la frequenza delle collisioni di testa negli sport di contatto come il football americano, la prevenzione del trauma cranico richiederà un cambiamento culturale nel modo in cui lo sport viene insegnato e praticato. Formazione per tecniche di pratica sicura, come placcaggio sicuro e colpire, mentre penalizzando i colpi sconsiderati offrirà benefici misurabili. Ulteriori cambiamenti devono includere la creazione di un ambiente di sicurezza, in cui i giocatori sono incoraggiati a segnalare i sintomi agli arbitri, allenatori e ai medici di squadra. Inoltre, stabilire un profilo neurocognitivo di base potrebbe essere usato come un marcatore di riferimento clinico per monitorare i cambiamenti nella presentazione neuropsichiatrica dei giocatori. Spetta ai medici della squadra rimuovere dal campo i giocatori che hanno subito anche un lieve TBI non complicato per ulteriori valutazioni (32).
Ci sono una serie di sfide esistenti relative alla CTE da affrontare. Anche se l’incidenza della commozione cerebrale legata allo sport è stato segnalato per variare da 1,6 milioni a 3,8 milioni, l’incidenza e la prevalenza di CTE rimane in gran parte sconosciuto (33). Una spiegazione di questo vuoto di conoscenza è forse dovuta al fatto che gli atleti esposti a colpi subconcussivi cumulativi, che esercitano una forza sufficiente a conferire danni neuronali ma inizialmente non hanno sintomi clinici evidenti, spesso non sono valutati o diagnosticati in modo tempestivo (34). Studi prospettici su larga scala, come il monitoraggio degli atleti con TBI multipli per un periodo predefinito, aggiungerebbero alla nostra comprensione del corso naturale e della fenomenologia della malattia. Il CTE è sempre più sotto i riflettori del pubblico attraverso i mass media. Sono necessari continui sforzi per diagnosticare, valutare e trattare questa malattia devastante. Il progresso esponenziale delle tecniche di neuroimaging e la comprensione dei meccanismi neuropatologici della malattia porteranno ad una diagnosi più precoce e ad interventi di trattamento tempestivi.
Contributi dell’autore
L’autore conferma di essere l’unico contributore di questo lavoro e di averlo approvato per la pubblicazione.
Dichiarazione di conflitto di interessi
L’autore dichiara che la ricerca è stata condotta in assenza di relazioni commerciali o finanziarie che potrebbero essere interpretate come un potenziale conflitto di interessi.
Riconoscimenti
L’autore riconosce il grande supporto, la guida e la mentorship del Dr. Stephen Strittmatter.
1. McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, et al. encefalopatia traumatica cronica negli atleti: tauopatia progressiva dopo trauma cranico ripetitivo. J Neuropathol Exp Neurol. (2009) 68:709-35. doi: 10.1097/NEN.0b013e3181a9d503
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, et al. encefalopatia traumatica cronica in veterani militari esposti all’esplosione e un modello di topo neurotrauma esplosione. Sci Transl Med. (2012) 4:134ra60. doi: 10.1126/scitranslmed.3003716
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Martland H. Dementia pugilistica. JAMA. (1928) 91:364-65. doi: 10.1001/jama.1928.02700150029009
CrossRef Full Text | Google Scholar
4. Millspaugh J. Dementia pugilistica. US Naval Med Bull. (1937) 35:303.
Google Scholar
5. Critchley M. Hommage a Clovis Vincent. Parigi: Maloine (1949).
Google Scholar
6. Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, et al. Valutazione clinicopatologica di encefalopatia traumatica cronica in giocatori di football americano. JAMA. (2017) 318:360-70. doi: 10.1001/jama.2017.8334
PubMed Abstract | CrossRef Full Text | Google Scholar
7. McKee AC, Stein TD, Kiernan PT, Alvarez VE. La neuropatologia dell’encefalopatia traumatica cronica. Brain Pathol. (2015) 25:350-64. doi: 10.1111/bpa.12248
PubMed Abstract | CrossRef Full Text | Google Scholar
8. McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, et al. Il primo incontro di consenso NINDS/NIBIB per definire i criteri neuropatologici per la diagnosi di encefalopatia cronica traumatica. Acta Neuropathol. (2016) 131:75-86. doi: 10.1007/s00401-015-1515-z
PubMed Abstract | CrossRef Full Text | Google Scholar
9. McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, et al. Lo spettro della malattia in encefalopatia cronica traumatica. Cervello. (2013) 136:43-64. doi: 10.1093/brain/aws307
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Jordan BD. Lo spettro clinico di lesioni cerebrali traumatiche legate allo sport. Nat Rev Neurol. (2013) 9:222-30. doi: 10.1038/nrneurol.2013.33
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Stern RA, Daneshvar DH, Baugh CM, Seichepine DR, Montenigro PH, Riley DO, et al. Presentazione clinica di encefalopatia traumatica cronica. Neurologia. (2013) 81:1122-9. doi: 10.1212/WNL.0b013e3182a55f7f
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Gardner A, Iverson GL, McCrory P. Encefalopatia cronica traumatica nello sport: una revisione sistematica. Br J Sports Med. (2014) 48:84-90. doi: 10.1136/bjsports-2013-092646
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Montenigro PH, Baugh CM, Daneshvar DH, Mez J, Budson AE, Au R, et al. Sottotipi clinici di encefalopatia traumatica cronica: revisione della letteratura e criteri diagnostici di ricerca proposti per la sindrome di encefalopatia traumatica. Alzheim Res Ther. (2014) 6:68. doi: 10.1186/s13195-014-0068-z
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Mawdsley C, Ferguson F. Malattia neurologica nei pugili. Lancet. (1963) 282:795-801. doi: 10.1016/S0140-6736(63)90498-7
CrossRef Full Text | Google Scholar
15. Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH. Encefalopatia traumatica cronica in un giocatore della National Football League. Neurochirurgia. (2005) 57:128-34. doi: 10.1227/01.NEU.0000163407.92769.ED
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensibilità dei criteri diagnostici rivisti per la variante comportamentale della demenza frontotemporale. Cervello. (2011) 134:2456-77. doi: 10.1093/brain/awr179
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Omalu BI, Bailes J, Hammers JL, Fitzsimmons RP. Encefalopatia traumatica cronica, suicidi e parasuicidi in atleti professionisti americani: il ruolo del patologo forense. Am J Forensic Med Pathol. (2010) 31:130-2. doi: 10.1097/PAF.0b013e3181ca7f35
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Maroon JC, Winkelman R, Bost J, Amos A, Mathyssek C, Miele V. Encefalopatia traumatica cronica negli sport di contatto: una revisione sistematica di tutti i casi patologici riportati. PLoS ONE. (2015) 10:e0117338. doi: 10.1371/journal.pone.0117338
CrossRef Full Text | Google Scholar
19. Iverson GL. Encefalopatia traumatica cronica e rischio di suicidio negli ex atleti. Br J Sports Med. (2014) 48:162-4. doi: 10.1136/bjsports-2013-092935
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Iverson GL, Gardner AJ, McCrory P, Zafonte R, Castellani RJ.Iverson Una revisione critica di encefalopatia traumatica cronica. Neurosci Biobehav Rev. (2015) 56:276-93. doi: 10.1016/j.neubiorev.2015.05.008
CrossRef Full Text | Google Scholar
21. Randolph C. L’encefalopatia cronica traumatica non è una vera malattia. Arch Clin Neuropsychol. (2018) 33:644-8. doi: 10.1093/arclin/acy063
CrossRef Full Text | Google Scholar
22. Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R, et al. Novel tau filament fold in encefalopatia traumatica cronica racchiude molecole idrofobiche. Nature. (2019) 568:420-3. doi: 10.1038/s41586-019-1026-5
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Weir DR, Jackson JS, Sonnega A, et al. National Football League Player Care Foundation Study of Retired NFL Players. Ann Arbor: University of Michigan Institute for Social Research (2009).
Google Scholar
24. King NS, Kirwilliam S. Permanenti sintomi post-concussivi dopo lieve trauma cranico. Brain Inj. (2011) 25:462-70. doi: 10.3109/02699052.2011.558042
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Ward A, Crean S, Mercaldi CJ, Collins JM, Boyd D, Cook MN, et al. Prevalenza del genotipo apolipoproteina E4 e omozigoti (APOE e4/4) tra i pazienti con diagnosi di malattia di Alzheimer: una revisione sistematica e meta-analisi. Neuroepidemiologia. (2012) 38:1-17. doi: 10.1159/000334607
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Jordan BD, Relkin NR, Ravdin LD, Jacobs AR, Bennett A, Gandy S. Apolipoproteina Eε4 associato con lesioni cerebrali traumatiche croniche nella boxe. (1997) 278:136-40. doi: 10.1001/jama.278.2.136
CrossRef Full Text | Google Scholar
27. Stern Y. Riserva cognitiva nell’invecchiamento e nella malattia di Alzheimer. Lancet Neurol. (2012) 11:1006-12. doi: 10.1016/S1474-4422(12)70191-6
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Laurent CL, Buée L, Blum D. Tau e neuroinfiammazione: Quale impatto per la malattia di Alzheimer e le tauopatie? Biomed J. (2018) 41:21-33. doi: 10.1016/j.bj.2018.01.003
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Sangobowale MA, Grin’kina NM, Whitney K, Nikulina E, St., Laurent-Ariot K, Ho JS, et al. Minociclina più N-acetilcisteina ridurre i deficit comportamentali e migliorare istologia con una finestra temporale clinicamente utile. J Neurotrauma. (2018) 35:907-17. doi: 10.1089/neu.2017.5348
CrossRef Full Text | Google Scholar
30. Haber M, James J, Kim J, Sangobowale M, Irizarry R, Ho J, et al. Minociclina più N-acteilcisteina induce rimielinizzazione, protegge sinergicamente oligodendrociti e modifica la neuroinfiammazione in un modello di ratto di lieve lesione cerebrale traumatica. J Cereb Blood Flow Metab. (2018) 38:1312-6. doi: 10.1177/0271678X17718106
PubMed Abstract | CrossRef Full Text | Google Scholar
31. Livingston G, Sommerlad A, Orgeta V, Costafreda SG, Huntley J, Ames D, et al. Dementia prevenzione, intervento e cura. Lancet. (2017) 390: 2673-734. doi: 10.1016/S0140-6736(17)31363-6
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Saulle M, Greenwald BD. Encefalopatia traumatica cronica: una revisione. Rehabil Res Pract. (2012). 2012: 816069. doi: 10.1155/2012/816069
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Langlois JA, Rutland-Brown W, Wald MM. L’epidemiologia e l’impatto delle lesioni cerebrali traumatiche. J Head Trauma Rehabil. (2006) 21:375-8. doi: 10.1097/00001199-200609000-00001
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Martini D, Eckner J, Kutcher J, Broglio SP. Biomeccanica di impatto subconcussivo della testa: confrontando diversi schemi offensivi. Med Sci Sport Exerc. (2013) 45:755-61. doi: 10.1249/MSS.0b013e3182798758
PubMed Abstract | CrossRef Full Text | Google Scholar