US Pharm. 2012;37(5):HS-2-HS-7.
La douleur est la raison la plus courante pour laquelle les gens consultent un médecin.1Une étude récente estime que la douleur affecte des dizaines de millions d’Américains et coûte aux États-Unis 635 milliards de dollars par an en raison de la consommation accrue de soins médicaux et des coûts indirects liés aux journées de travail manquées et à la perte de productivité.2 La prise en charge de la douleur chronique avec des médicaments tels que les opioïdes est une stratégie courante, mais la douleur chronique peut persister ou s’aggraver malgré un traitement opioïde agressif. Cet article vise à mettre en lumière le phénomène des opioïdes prescrits pour traiter la douleur, mais qui provoquent une douleur nouvelle ou paradoxalement aggravée, appelée hyperalgésie induite par les opioïdes (OIH). Cette condition est appelée hyperalgésie induite par les opioïdes (OIH). L’OIH peut être plus formellement définie comme une sensibilisation nociceptive accrue causée par l’exposition aux opioïdes.3 L’OIH diffère distinctement de la tolérance, de l’accoutumance, de la dépendance et de la progression de la maladie. La prévalence clinique de l’HIO est inconnue.4
Diagnostic et présentation
L’un des principaux problèmes pour diagnostiquer correctement l’HIO est la ressemblance étroite de cette affection avec la tolérance aux opiacés. La tolérance et l’HSO partagent la caractéristique d’une réponse analgésique réduite à la dose d’opioïdes, et il est probable qu’elles partagent bon nombre des mêmes mécanismes cellulaires.5 La tolérance peut être surmontée en augmentant la dose d’opioïdes, alors que la même augmentation chez un patient atteint d’HSO entraîne une aggravation de la douleur.4 De plus, la tolérance a tendance à se développer lentement dans le temps, alors que l’augmentation de la douleur résultant du traitement aux opioïdes chez le patient atteint d’HSO se produit relativement rapidement. Souvent, l’OIH diffère de la tolérance en ce sens que l’intensité de la douleur est plus forte que ce qui avait été initialement signalé.6
La douleur non traitée est une autre possibilité qui doit être écartée. Si le traitement opioïde est sous-optimal, l’augmentation de la dose devrait conduire à un soulagement de la douleur ; si l’HIO est présente, le contraire devrait être vrai. Pour que le diagnostic de l’OIH soit exact, la douleur doit disparaître après l’arrêt du traitement par l’opioïde en cause.6 Pour compliquer encore les choses, l’hyperalgésie résultant du sevrage des opioïdes est un phénomène bien documenté.7 Ainsi, la disparition de la douleur après l’arrêt des opioïdes dans le cas de l’OIH ne sera pas immédiate et nécessitera de la patience. L’OIH tend à présenter d’autres caractéristiques distinctes (TABLEAU 1). La douleur associée à l’OIH tend à être plus diffuse et de moindre qualité, et les stimuli nocifs tendent à être plus douloureux que ce à quoi on pourrait normalement s’attendre. Dans l’OIH, la douleur se manifeste souvent dans des zones s’étendant au-delà de la région de la blessure ou de la lésion tissulaire. La douleur peut persister dans l’OIH malgré l’élimination de la source initiale de la douleur ou la guérison des tissus endommagés. Dans l’OIH, au fur et à mesure que le traitement opioïde progresse, la douleur peut donner l’illusion de s’aggraver par rapport à ce qu’elle était au départ, malgré le temps, le repos et d’autres mesures qui permettraient normalement une guérison cliniquement pertinente5. De plus, l’allodynie a été démontrée dans un certain nombre d’études humaines et animales de l’OIH.8

Etiologie
Il existe un certain nombre de théories concernant la cause de l’OIH. La théorie qui a fait l’objet du plus grand nombre de recherches et qui reçoit actuellement le plus d’attention est le modèle neuroexcitateur. On pense que certains opioïdes et leurs métabolites agonisent le récepteur N-méthyl-D-aspartate (NMDA). L’activation du récepteur NMDA provoque un afflux de calcium qui augmente considérablement l’excitabilité du neurone. Lorsque le récepteur NMDA et les neurones correspondants sont plus actifs, ils peuvent transmettre plus facilement les impulsions douloureuses initiées par la substance circulante ou d’autres stimuli nocifs. Le glutamate est le principal agoniste endogène du récepteur NMDA.9
Les études montrant un soulagement de l’OIH après l’administration d’antagonistes des récepteurs NMDA soutiennent cette théorie de la pronociception modulée par l’activation des récepteurs NMDA. Ces antagonistes se lient de manière non compétitive au site de la phencyclidine sur le canal NMDA et bloquent l’influx de calcium qui se produirait lorsque le récepteur se lie au glutamate ou à un autreagoniste.10 Des études animales et humaines ont démontré que les sujets atteints d’hyperactivité vésicale auxquels on avait administré de la kétamine, un antagoniste des récepteurs NMDA, obtenaient de meilleurs résultats lors de tests contrôlés de stimulation de la douleur. Dans une étude, des rats ont reçu des injections intrathécales de morphine pendant plusieurs jours jusqu’à ce qu’ils présentent une OIH sensible à la chaleur. L’administration de dizocilpine, un inhibiteur du NMDA, a été efficace pour inverser, au moins partiellement, l’hyperalgésie thermique5. D’autres antagonistes des récepteurs NMDA, comme le dextrométhorphane, ont montré qu’ils pouvaient soulager les états hyperalgésiques par l’inhibition des récepteurs NMDA.
Les opiacés, comme la codéine, l’hydromorphone et la morphine, subissent un certain nombre de biotransformations dans le cadre du processus métabolique normal.La codéine est métabolisée en morphine par l’enzyme CYP2D6. La codéine est métabolisée en morphine par l’enzyme CYP2D6. La morphine est métabolisée en un certain nombre de molécules, mais elle est principalement métabolisée par glucuronidation en morphine-3-glucuronide (M3G) et, dans une moindre mesure, en morphine-6-glucuronide (M6G) (FIGURE 1).11
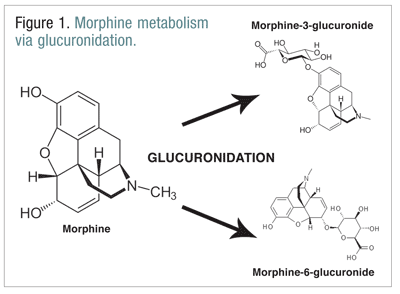
Le M6G est un métabolite thérapeutiquement important. Il a une affinité de liaison aux récepteurs mu similaire à celle de la morphine et est capable de produire une analgésie, une dépression respiratoire et d’autres effets similaires à ceux de la morphine. Le M3G n’est cependant pas un agoniste du récepteur mu, et son affinité pour le récepteur mu est faible par rapport à celle de la morphine. Contrairement à la morphine, au M6G et aux autres opioïdes, le M3G est un agoniste du NMDA. Par glucuronidation, le M3G est converti à un taux environ six fois supérieur à celui du M6G.12 D’autres opioïdes ont été impliqués dans une activité agoniste NMDA et des effets neuroexcitateurs paradoxaux.6
La preuve que l’OIH n’est pas soulagée par l’administration d’un antagoniste opioïde (par exemple, la naloxone) appuie la théorie selon laquelle l’OIH est causée par l’activité de récepteurs autres que le récepteur opioïde mu. Chez les patients qui ont expérimenté l’OIH alors qu’ils étaient sous traitement opioïde à haute dose, l’administration de naloxone a provoqué une aggravation de la douleur.13 En déplaçant l’opioïde du récepteur mu, la naloxone a empêché l’opioïde de fournir l’analgésie qui était possible par l’activation du récepteur mu, alors que, simultanément, l’hyperalgésie n’a pas été remise en cause14.
La dynorphine peut également être impliquée dans le développement de l’OIH.15La dynorphine est un peptide opioïde endogène qui se lie principalement au récepteur opioïde kappa et, dans une moindre mesure, aux récepteurs opioïdes mu et NMDA. On pense que l’agonisme de la dynorphine sur les récepteurs opioïdes kappa et les récepteurs NMDA joue un rôle dans la pronociception de l’OIH malgré la présence d’agonistes purs des récepteurs mu. Des études ont montré que l’inversion de la dynorphine peut restaurer les effets analgésiques de la morphine lorsque les sujets reçoivent un antisérum de la dynorphine. En outre, on sait que la dynorphine augmente lors d’une administration prolongée d’opioïdes. La dynorphine provoque également la libération de neuropeptides excitateurs dans différentes localisations du système nerveux central.
Options de traitement
Une fois le diagnostic d’OIH posé, il existe un certain nombre d’options de traitement parmi lesquelles il faut soigneusement choisir. Si la blessure douloureuse initiale ou la lésion tissulaire a disparu et que la douleur persiste malgré et à cause du traitement opioïde, l’approche la plus directe consiste à arrêter l’opioïde en cause. Cela doit être fait progressivement pour minimiser les effets indésirables du sevrage. Il faut noter que l’hyperalgésie peut probablement s’aggraver au début du processus d’arrêt.16 Cela présente une situation éthique difficile dans laquelle le clinicien peut avoir des difficultés à convaincre le patient que le médicament prescrit pour traiter la douleur peut avoir causé ou aggravé la douleur et que la douleur peut encore s’aggraver avant de disparaître.17
Si la douleur légitime persiste et qu’une certaine quantité d’analgésie avec des opioïdes est nécessaire, d’autres stratégies que l’arrêt total des opioïdes doivent être explorées. Les patients souffrant d’OIH peuvent être soulagés en réduisant la dose d’opioïdes.4 On a rapporté que des patients ont trouvé un équilibre acceptable entre l’analgésie et le soulagement de l’hyperalgésie en réduisant la dose d’opioïdes.
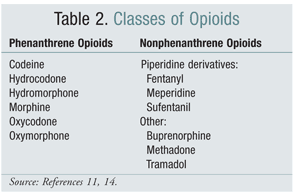
Le passage d’une classe structurelle d’opioïdes à une autre a été une option efficace pour atténuer l’OIH dans certaines études. Les études menées à ce jour ont démontré que l’HIO est plus fortement associée aux opioïdes de la classe des phénanthrènes (TABLEAU 2).4 La titration de l’opioïde phénanthrène et la conversion à un autre opioïde peuvent permettre de résoudre l’HIO. La codéine, l’hydromorphone, la morphine et les opioïdes de structure similaire subissent une glucuronidation dans le cadre de leur métabolisme. Il est possible d’éviter la présence d’un métabolite glucuronide activant les récepteurs de la NMDA en passant à un opioïde dont la structure est unique, comme le fentanyl. Au moment où nous écrivons ces lignes, l’HSO n’a pas été démontrée dans les essais impliquant l’oxymorphone.4
Supplémenter le traitement opioïde avec un inhibiteur de la cyclo-oxygénase 2 (COX-2) est une autre stratégie qui bénéficie d’un certain soutien. En réduisant la synthèse des prostaglandines, les inhibiteurs de la COX-2 peuvent diminuer la sensibilisation des neurones pronociceptifs. Les anti-inflammatoires non stéroïdiens (AINS) et les inhibiteurs de la COX-2 semblent avoir des effets analgésiques indépendants de leur capacité à supprimer la synthèse des prostaglandines au niveau périphérique.9 Les inhibiteurs de la COX-2 ont également démontré leur capacité à antagoniser le récepteur NMDA. Au niveau central, les AINS sont capables d’antagoniser le récepteur NMDA en bloquant le glutamate, la substance P et d’autres acides aminés excitateurs. Indépendamment de cela, l’activation du récepteur NMDA peut réguler à la hausse l’expression de la COX-2.18
Etant donné l’attention que le récepteur NMDA a reçu pour son rôle dans l’HIO, l’antagonisme de ce récepteur semblerait être une stratégie de traitement raisonnable. La kétamine a fait l’objet de nombreuses recherches, mais ses effets indésirables peuvent être graves. Bien qu’elle antagonise le NMDA et fournisse un effet anesthésique qui favorise l’analgésie, les effets secondaires sont courants, divers et graves.19 Ces effets comprennent la tachyarythmie, l’hypertension, les troubles cognitifs et les réactions psychomimétiques, notamment les changements d’humeur, les rêves vifs, le délire, les hallucinations et la sédation. La kétamine est efficace pour inverser l’hyperalgésie et augmenter les effets des opioïdes chez les patients recevant de fortes doses, mais ses effets indésirables l’empêchent d’être une option thérapeutique viable.
Le dextrométhorphane est un autre antagoniste des récepteurs NMDA qui a été étudié pour le traitement de l’OIH. Un produit combiné contenant un mélange 1:1 de morphine et de dextrométhorphane a été étudié.20 Les études n’ont pas réussi à démontrer que la thérapie combinée pouvait atteindre une analgésie supérieure à celle de la morphine seule. Ces études ont été critiquées parce que la quantité de dextrométhorphane utilisée était insuffisante pour antagoniser adéquatement les récepteurs NMDA21.On ne connaît toujours pas la dose de dextrométhorphane nécessaire pour soit augmenter l’analgésie d’un opioïde, soit prévenir l’OIH causée par l’activation des récepteurs NMDA.
La méthadone est un opioïde aux qualités uniques qui en font une option convaincante pour traiter la douleur chez le patient souffrant d’OIH. En plus de ses effets analgésiques par la liaison au récepteur opioïde mu, la méthadone est un faible antagoniste des récepteurs NMDA.4 Il a été rapporté que l’ajout d’une dose même faible de méthadone a été efficace pour réduire l’hyperalgésie. Dans un cas, l’ajout d’une faible dose de méthadone (10 mg deux fois par jour) a amélioré les rapports de douleur de façon marquée, avec une réduction de la dose totale d’opioïdes de 40 à 50 %.22 Le passage de l’opioïde en cause à la méthadone a été une stratégie de traitement populaire. Le tramadol et la mépéridine ont également une certaine activité antagoniste des récepteurs NMDA.
Une autre modalité de traitement qui a un certain potentiel pour traiter l’HSO est la buprénorphine.23,24 La buprénorphine est un agoniste opioïde partiel au niveau du récepteur mu et a également une activité antagoniste au niveau du récepteur kappa.4La dynorphine, un agoniste du récepteur kappa, est connue pour augmenter pendant le traitement avec des opioïdes. L’activation du récepteur kappa antagonise en grande partie les effets des opioïdes médiés par le récepteur mu.25 L’utilité clinique de la buprénorphine dans le traitement de l’OIH n’a pas encore été complètement expliquée.
Conclusion : Perspectives d’avenir
Il reste beaucoup de travail à faire avant de pouvoir déclarer que l’OIH est comprise. Le mécanisme cellulaire exact et les voies de signalisationresponsables de ce phénomène ne sont pas définis. Pour compliquer la situation, il existe des rapports contradictoires sur le rôle clé ou non de certains composants. L’essentiel de la recherche sur l’élucidation de la physiopathologie de l’OIH a porté sur le modèle animal. Cela a une certaine valeur, mais les informations provenant du modèle humain sont rares.
Une faible compréhension de la cause d’une affection génère un arsenal incertain d’options thérapeutiques. La meilleure stratégie disponible est l’arrêt total de l’opioïde problématique, mais elle est peu utile pour le patient qui a besoin d’un soulagement de la douleur de type opioïde.
Plusieurs autres modalités de traitement ont été explorées, mais toutes bénéficieraient d’une étude plus approfondie. La majorité des études réalisées chez l’homme concernent des patients en phase périsurgicale, des toxicomanes en voie de guérison et des modèles expérimentaux de douleur. Ces études ne sont pas sans valeur, mais ces populations ne reflètent pas la population souffrant de douleur chronique. Les autres limites des options de traitement disponibles comprennent des résultats peu encourageants, une toxicité excessive malgré un succès modéré et des études de petite taille produisant des conclusions faibles.
À ce jour, un certain nombre d’études cliniques utilisant la mémantine, un antagoniste des récepteurs NMDA, pour traiter l’OIH sont en cours ou prévues.26 Il existe encore des rapports contradictoires quant au rôle exact des récepteurs NMDA dans l’OIH. Des recherches supplémentaires doivent être menées pour déterminer les récepteurs et les mécanismes responsables clés présents dans l’OIH. L’élargissement de la taille de l’échantillon des études permettra d’étayer les conclusions déjà formulées sur les agents thérapeutiques responsables de l’hyperactivité vésicale et efficaces pour la traiter. La réalisation d’études axées sur la douleur chronique, plutôt que sur des modèles de douleur aiguë, peut apporter des informations cliniquement pertinentes au praticien.
Chapman et ses collègues ont soulevé des questions spécifiques auxquelles il faut répondre avant de pouvoir diagnostiquer l’OIH et de pouvoir administrer un traitement efficace.27 Quelles sont les relations entre la dose et la durée des opioïdes et l’OIH ? Avec quel type de douleur, ou chez quel type de patient, l’HIO est-elle plus susceptible de se développer ? Ce sont les questions qui doivent guider les futures recherches cliniques.
1. Parselis Kelly J, Cook SF, Kaufman DW, et al. Prévalence et caractéristiques de l’utilisation des opioïdes dans la population adulte américaine. Pain. 2008 ;138:507-513.
2. Institut de médecine. Soulager la douleur en Amérique : Un plan directeur pour transformer la prévention, les soins, l’éducation et la recherche. Washington, DC : The National Academies Press ; 2011.
3. Chu LF, Angst MS, Clark D. Hyperalgésie induite par les opioïdes chez les humains : mécanismes moléculaires et considérations cliniques. Clin J Pain. 2008;24:479-496.
4. Lee M, Silverman SM, Hansen H, et al. A comprehensive review of opioid-induced hyperalgesia. Médecin de la douleur. 2011;14:145-161.
5. Mao J. Sensibilité anormale à la douleur induite par les opioïdes : implications dans la thérapie clinique des opioïdes. Pain. 2002;100:213-217.
6. Raffa RB, Pergolizzi JV Jr. L’hyperalgésie induite par les opioïdes : est-elle cliniquement pertinente pour le traitement des patients douloureux ? Pain Manage Nurs. 2011;1-17.
7. Li X, Clark JD. Hyperalgésie pendant l’abstinence d’opioïdes : médiation par le glutamate et la substance P. Anesth Analg. 2002;95:979-984.
8. Pud D, Cohen D, Lawental E, Eisenberg E. Opioids and abnormal painperception : new evidence from a study of chronic opioid addicts andhealthy subjects. Drug Alcohol Depend. 2006;82:218-223.
9. Malmberg AB, Yaksh TL. Hyperalgesia mediated by spinal glutamateor substance P receptor blocked by spinal cyclooxygenase inhibition. Science. 1992;257:1276 -1279.
10. Fisher K, Coderre TJ, Hagen NA. Targeting theN-methyl-D-aspartate receptor for chronic pain management. Preclinicalanimal studies, recent clinical experience and future researchdirections. J Pain Symptom Manage. 2000;20:358-373.
11. Lötsch J. Métabolites des opioïdes. J Pain Symptom Manage. 2005;29:S10-S24.
12. Skarke C, Lötsch J. Morphine metabolites : clinical implications. Semin Anesth, Perioperative Med Pain. 2002;21:258-264.
13. Juni A, Klein G, Kest B. Morphine hyperalgesia in mice isunrelated to opioid activity, analgesia, or tolerance : evidence formultiple diverse hyperalgesic systems. Brain Res. 2006;1070:35-44.
14. Angst M, Clark JD. Opioid-induced hyperalgesia : a qualitative systematic review. Anesthesiology. 2006;104:570-587.
15. Vanderah TW, Gardell LR, Burgess SE, et al. La dynorphine favorise la douleur anormale et la tolérance antinociceptive opioïde spinale. J Neurosci. 2000;20:7074-7079.
16. Hooten WM, Mantilla CB, Sandroni P, Townsend CO. Associationsbetween heat pain perception and opioid dose among patients with chronicpain undergoing opioid tapering. Pain Med. 2010;11:1587-1598.
17. Silverman S. Opioid induced hyperalgesia : clinical implications for the pain practitioner. Médecin de la douleur. 2009;12:679-684.
18. Li SQ, Xing YL, Chen WN, et al. L’activation du récepteur NMDA est associée à la régulation à la hausse de l’expression de COX-2 dans le cornet dorsal spinal pendant les entrées nociceptives chez les rats. Neurochem Res. 2009;34:1451-1463.
19. Okon T. Ketamine : une introduction pour le médecin de la douleur et de la médecine palliative. Pain Physician. 2007;10:493-500.
20. Galer BS, Lee D, Ma T, et al. MorphiDex (combinaison de morphinesulfate/dextrométhorphane hydrobromure) dans le traitement de la douleur chronique : trois essais cliniques multicentriques, randomisés, en double aveugle et contrôlés ne démontrent pas une analgésie opioïde accrue ou une réduction de la tolérance. Pain. 2005;115:284-295.
21. Wiesenfeld-Hallin Z. Thérapies combinées opioïde-antagoniste NMDA.Quels avantages offrent-elles pour le contrôle des syndromes douloureux ? Médicaments. 1998;55;1-4.
22. Vorobeychik Y, Chen L, Bush MC, Mao J. Improved opioid analgesic effect following opioid dose reduction. Pain Med. 2008;9:724-727.
23. Baron MJ, McDonald PW. Réduction significative de la douleur chez les patients souffrant de douleur chronique après la désintoxication des opioïdes à forte dose. J Opioid Manag. 2006;2:277-282.
24. Koppert W, Ihmsen H, Körber N, et al. Different profiles ofbuprenorphine-induced analgesia and antihyperalgesia in a human painmodel. Pain. 2005;118:15-22.
25. Pan ZZ. Actions mu-opposantes du récepteur kappa-opioïde. Trends Pharmacol Sci. 1998;19:94-98.
26. Ballantyne JC, Mao J. Opioid therapy for chronic pain. N Engl J Med. 2003;349:1943-1953.
27. Chapman CR, Lipschitz DL, Angst MS, et al. Opioid pharmacotherapyfor chronic non-cancer pain in the United States : a research guidelinefor developing an evidence-base. J Pain. 2010;11:807-829.