Chronic Traumatic Encephalopathy (CTE) jest charakterystyczną chorobą neurodegeneracyjną związaną z białkiem tau. Obserwuje się wzrost liczby rozpoznań CTE u sportowców, zwłaszcza zawodników futbolu amerykańskiego, jak również u weteranów wojskowych w warunkach bojowych (1, 2). Chociaż CTE zostało publicznie rozpoznane stosunkowo niedawno, po raz pierwszy opisano je jako zespół „punch drunk” w klasycznym artykule Martlanda i wsp. (3). Raport dotyczył kilku bokserów, którzy w trakcie swojej kariery zawodowej doznawali powtarzających się uderzeń głową i prezentowali zarówno objawy psychiatryczne, jak i poważne deficyty pamięciowe i neurokognitywne, analogiczne do typowych pacjentów z demencją (3). Nomenklatura choroby ewoluowała do „dementia pugilistica” (4), a ostatecznie do CTE w 1949 roku (5).
CTE ma unikalną charakterystykę neuropatologiczną, na którą składa się nagromadzenie fosforylowanego tau (p-tau) w bruzdach i regionach okołonaczyniowych, mikroglejoza i astrocytoza. Te zmiany patologiczne prowadzą do postępującej, wyniszczającej neurodegeneracji. Na podstawie schematu progresji patologicznej, CTE dzieli się na cztery odpowiednie stadia (Rycina 1). W I stadium CTE mózg wydaje się być normalny, ale p-tau znajduje się w ograniczonej liczbie miejsc, często w korze bocznej i czołowej, a także w pobliżu małych naczyń krwionośnych w głębi bruzd. W locus coeruleus może występować niewielka liczba splątków neurofibrylarnych (NFT) i neurytów. W stadium II można stwierdzić zlokalizowane makroskopowe nieprawidłowości. Na przekrojach anatomicznych i w badaniach neuroobrazowych obserwuje się powiększenie komór bocznych, cavum septum pellucidum z fenestracją lub bez fenestracji, a także bladość jądra miażdżystego i istoty czarnej. Występują liczne ogniska p-tau w głębi bruzd i pojawia się wzór rozsiewu. W stadium III większość wycinków patologicznych wykazuje nieprawidłowości makroskopowe. Występuje globalna utrata masy mózgu, łagodny zanik płatów czołowych i skroniowych oraz poszerzenie komór. U połowy pacjentów z CTE stwierdza się nieprawidłowości przegrody międzykomorowej, w tym cavum septum pellucidum. Patologia P-tau rozprzestrzenia się, obejmując korę czołową, skroniową, ciemieniową i wyspową. W stadium IV zmniejszenie masy mózgu jest dramatyczne, opisywano przypadki, w których masa mózgu wynosiła 1000 g (w porównaniu do 1300 – 1400 g w prawidłowych mózgach). Występuje głęboka atrofia płatów czołowych, skroniowych przyśrodkowych, a także przedniej części talii. Występuje również zanik dróg istoty białej. U większości pacjentów w stadium czwartym występują nieprawidłowości przegrody międzykomorowej. Rozprzestrzenianie się p-tau dotyczy większości regionów, w tym kory podskroniowej (7, 8). U większości pacjentów z CTE obserwuje się również nieprawidłowości w zakresie fosforylowanego białka wiążącego DNA TAR 43 kDa (TDP-43). Miąższowa patologia TDP-43 ma również charakter postępujący, podobny do anatomicznego wzorca rozprzestrzeniania się p-tau. Immunoreaktywność TDP-43 stwierdza się w prawie wszystkich przypadkach choroby w stadium IV (7).

Rysunek 1. Powyższe obrazy przedstawiają cztery stadia CTE wg McKee’go.
Fenotyp kliniczny CTE nie został jeszcze jednoznacznie zdefiniowany. Poniższe akapity przedstawiają próby charakterystyki objawów CTE w różnych stadiach procesu chorobowego (Tabela 1). Zgodnie z klasyfikacją McKee, w I stadium typowy pacjent z CTE jest bezobjawowy lub może skarżyć się na łagodne deficyty pamięci krótkotrwałej i objawy depresyjne. Może być obserwowana łagodna agresja. W stadium II, objawy nastroju i zachowania mogą obejmować wybuchy zachowań i bardziej nasilone objawy depresyjne. W stadium III pacjenci zwykle prezentują więcej deficytów poznawczych, w tym utratę pamięci, deficyty funkcjonowania wykonawczego, zaburzenia funkcji wzrokowo-przestrzennych i apatię. W stadium IV u pacjentów występują zaawansowane deficyty językowe, objawy psychotyczne, w tym paranoja, deficyty ruchowe i parkinsonizm.
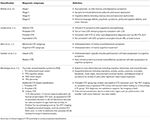
Tabela 1. Proponowane klasyfikacje kliniczne CTE.
Jordan i wsp. (10) byli jednymi z pierwszych, którzy scharakteryzowali klinicznie tę chorobę. Podzielili oni prezentacje kliniczne CTE na trzy domeny: behawioralną/psychiatryczną, poznawczą i motoryczną. Domena behawioralna i psychiatryczna obejmowała agresję, depresję, apatię, impulsywność, urojenia, w tym paranoję, i samobójstwo. Domena poznawcza obejmowała osłabienie uwagi i koncentracji, deficyty pamięci, deficyty funkcjonowania wykonawczego, zaburzenia funkcji wzrokowo-przestrzennych, deficyty językowe i otępienie. Wreszcie, cechy motoryczne obejmowały dyzartrię, zaburzenia chodu, ataksję i brak koordynacji, spastyczność oraz cechy parkinsonizmu, takie jak drżenie. Na podstawie tych cech klinicznych, jak również istniejących informacji neuropatologicznych, zdefiniowano cztery podtypy diagnostyczne, a mianowicie „Definitywne”, „Prawdopodobne”, „Możliwe” i „Nieprawdopodobne” CTE.
Stern i wsp. (11), oraz związane z nimi opisy przypadków (14, 15), różnili się w opisie typowego pacjenta z CTE, dzieląc prezentację kliniczną na dwa odrębne podtypy. W pierwszym podtypie występowały głównie zmiany zachowania i nastroju, a w drugim głównie zaburzenia funkcji poznawczych. U zdecydowanej większości pacjentów z podtypem zaburzeń nastroju i zachowania w miarę postępu choroby dochodziło do rozwoju deficytów poznawczych. Natomiast stosunkowo niewielu pacjentów z grupy poznawczej wykazywało zmiany nastroju lub zachowania w trakcie trwania choroby. W badaniu przeprowadzonym przez Sterna i wsp. (11) pacjenci z grupy poznawczej mieli znacząco wyższe prawdopodobieństwo rozwoju demencji. Byli oni również istotnie starsi w momencie postawienia diagnozy w porównaniu z pacjentami z grupy nastroju/zachowania. Podgrupa behawioralna pacjentów z CTE może przypominać pacjentów cierpiących na behawioralną odmianę otępienia czołowo-skroniowego (behavioral variant frontotemporal dementia – bvFTD), co sprawia, że diagnoza kliniczna jest trudniejsza. Jednak typowe dla bvFTD objawy behawioralne, takie jak apatia i zahamowanie, często nie są obserwowane u pacjentów z CTE (11, 16). Biorąc pod uwagę nieodłączną heterogenność bvFTD, jak również podobny charakter tauopatyczny obu chorób, odróżnienie bvFTD od CTE stanowi wyzwanie diagnostyczne.
Oprócz objawów behawioralnych CTE, związek między samobójstwem a CTE pozostaje tematem analizowanym w literaturze. Wcześniejsze badania, takie jak seria pięciu zawodowych sportowców z potwierdzonym rozpoznaniem CTE opisana przez Omalu i wsp. (17), sugerowały silny związek między CTE a samobójstwem. Autorzy sugerowali ponadto, że etiologia zachowań samobójczych/parauzji w populacji CTE może być częściowo spowodowana tauopatią w postaci splątków neurofibrylarnych i nici neurytowych w strategicznych jądrach mózgu limbicznego, takich jak locus ceruleus. Maroon i wsp. (18), dokonali przeglądu 153 potwierdzonych patologicznie przypadków CTE opublikowanych w latach 1954-2013. Odnotowali, że częstość występowania samobójstw w populacji CTE i przypadkowych zgonów wynosiła 11,7 i 17,5%, co znacznie przewyższało poziom populacji ogólnej wynoszący odpowiednio 1,5 i 4,8% (18). Zwolennicy przeciwnego poglądu sugerują, że samobójstwa były zgłaszane głównie we wcześniejszych stadiach CTE, a związek między postępem choroby a samobójstwami pozostaje obecnie niejasny (19).
W metaanalizie 158 przypadków przeprowadzonej przez Gardnera i wsp. (12) objawy kliniczne CTE zostały podzielone na „klasyczne” i „nowoczesne” objawy CTE, aby rozróżnić starszy opis przypadków CTE skoncentrowany głównie na bokserach w porównaniu z bardziej rozwiniętym opisem klinicznym, który dotyczy również zawodowych graczy futbolu amerykańskiego. Podczas gdy „klasyczne” objawy CTE zazwyczaj obejmowały dyzartrię, trudności w poruszaniu się, a następnie postępujące deficyty pamięci, „nowoczesny” obraz CTE obejmował również objawy neuropsychiatryczne, takie jak objawy depresyjne, paranoja, wycofanie społeczne i izolacja, zaburzona ocena sytuacji i agresja. Deficyty poznawcze, takie jak zanik pamięci, zaburzenia funkcji wykonawczych, zaburzenia językowe i przetwarzania informacji pojawiają się w późniejszym okresie procesu chorobowego (12).
Ponieważ definicja CTE zależy przede wszystkim od cech patologicznych, zaproponowano alternatywne określenie kliniczne zespołu encefalopatii pourazowej (traumatic encephalopathy syndrome – TES) autorstwa Montenigro i wsp. (13), opisujące kliniczne następstwa powtarzających się TBI. Autorzy oparli tę klasyfikację na przeglądzie 202 opublikowanych przypadków. TES jest bardziej wszechstronną diagnozą i może być podzielona na cztery podkategorie, w tym wariant behawioralny/nastrojowy TES, wariant poznawczy TES, wariant mieszany TES i otępienie TES. Proponowana diagnoza TES została oparta na istnieniu pięciu ogólnych kryteriów, trzech podstawowych cech klinicznych i dziewięciu cech pomocniczych. Wykorzystując istniejące biomarkery* (Tabela 1), zaproponowano dodatkowe kwalifikatory diagnostyczne, które obejmowały „prawdopodobne”, „możliwe” i „mało prawdopodobne” CTE (9, 13). Proponowana diagnoza TES również zawierała kwalifikatory czasowe i obejmowała „przebieg postępujący”, „przebieg stabilny” i „przebieg nieznany/niespójny”. Jeśli prezentacja kliniczna obejmowała również objawy motoryczne, takie jak parkinsonizm, dodawano również modyfikator „z cechami motorycznymi”.
W miarę jak rośnie nasze zrozumienie CTE, istnieje szereg wyzwań i krytyk, którymi należy się zająć. Jedną z hipotez, jako alternatywę dla zjawiska CTE, jest teoria zmniejszonej „rezerwy poznawczej”. Teoria ta mówi, że powtarzające się neurotraumy prowadzą do zmniejszenia rezerwy poznawczej i przyspieszenia rozwoju leżących u ich podłoża zaburzeń neurodegeneracyjnych (20, 21). Jeśli teoria ta byłaby prawdziwa, sugerowałaby, że CTE i AD znajdują się w tym samym spektrum neuropatologicznym. Twierdzenie to zasługuje na dalszą analizę. Podobnie jak w AD, izoformy Tau w CTE również składają się z mieszaniny izoform trzypowtórzeniowej (3R) i czteropowtórzeniowej (4R). Jednakże, zgodnie z ostatnim doniesieniem Falcona i wsp. (22), filamenty tau wyodrębnione z mózgów pacjentów z CTE zawierają również unikalny region ß-helisy z hydrofobowym wgłębieniem, który nie występuje w mózgach pacjentów z AD. Jama ta zawiera dodatkowy kofaktor, który, jak się uważa, może odgrywać funkcjonalną rolę w propagacji tau. Falcon i wsp. (22) sugerują, że lokalizacja inkluzji tau w pobliżu naczyń krwionośnych sugeruje, że kofaktory niezbędne do składania tau mogą przekraczać barierę krew-mózg po urazie głowy. Autorzy twierdzą również, że fakt, iż uraz mózgu prowadzi do CTE tylko w podgrupie osób, które doznały urazu, może być związany z wyższym poziomem kofaktorów u osób bardziej podatnych. Te kofaktory mogą stanowić cel terapeutyczny dla zapobiegania montażu tau i rozwoju CTE po urazie (22).
An alternative theory proposes that the psychiatric symptoms such as depression and anger reported in CTE patients are independent of the CTE disease process and are reported in a cofounded fashion. Zwolennicy tej hipotezy powołują się na wcześniejsze badania, takie jak to opisane przez Weir et al. (23), w którym 1,063 byłych graczy NFL zapytano, czy doświadczyli napadów złości. Stwierdzono, że 30,7% zawodników w wieku 30-49 lat i 29,3% zawodników w wieku 50 lat i starszych zgłaszało napady złości. Jednak autorzy zauważyli również, że zgłoszone miary gniewu były rzeczywiście niższe niż te zgłoszone dla ogólnej populacji USA, które wynosiły 54,8% dla mężczyzn w wieku od 30 do 49 lat i 47,2% dla mężczyzn w wieku powyżej 50 lat (23). Chociaż argumenty dotyczące współwystępowania objawów psychiatrycznych i chorób neurodegeneracyjnych, takich jak CTE, są trudne do zweryfikowania na podstawie wyników badań neuroobrazowych i neuropatologicznych, można zastosować podobne argumenty do objawów psychiatrycznych w każdej chorobie neurodegeneracyjnej, takiej jak AD, bvFTD, choroba Parkinsona (PD) lub stwardnienie zanikowe boczne (ALS).
Innym ważnym źródłem diagnostycznego zamieszania jest kliniczne rozgraniczenie między CTE a przedłużonym zespołem po wstrząśnieniu mózgu (PCS), zwłaszcza biorąc pod uwagę wcześniejsze doniesienia wskazujące, że ~10-20% osób, które doznały wstrząśnienia mózgu, doświadcza przedłużonych objawów. Przewlekły zespół pouderzeniowy (Chronic Postconcussive Syndrome – CPCS) odnosi się do utrzymywania się objawów PCS prowadzących do upośledzenia funkcjonowania, a często także sprawności sportowej, trwających dłużej niż 1 rok. Objawy CPCS obejmują bóle i zawroty głowy, zaburzenia uwagi, pamięci i funkcjonowania wykonawczego, objawy depresji i drażliwości (10). King i Kirkwilliam określili termin „trwałe PCS” w odniesieniu do osób z objawami PCS utrzymującymi się średnio 6,9 roku po wstrząśnieniu mózgu. Ponadto stwierdzili, że u znacznej liczby pacjentów z trwałym PCS (40-59%) występowały również przedchorobowe lub pozarobowe zaburzenia neuropsychiatryczne, takie jak depresja, lęk, PTSD i/lub ból (24). Jak twierdzą Jordan i wsp. (10), CPCS można klinicznie odróżnić od CTE na podstawie związku czasowego z ostrym zdarzeniem wstrząsowym. Dokładny i precyzyjny wywiad czasowy pozostaje kluczem do oceny neurologicznej. Co więcej, ból głowy jest główną cechą CPCS, ale nie jest często opisywany w CTE. Chociaż można się z tym spierać, u pacjentów w stadium I i II wg McKee mogą występować bóle głowy, co dodatkowo zwiększa złożoność możliwego nakładania się CTE & CPCS (9). Rozpoznanie CPCS pozostaje kontrowersyjne, ponieważ nie jest jasne, czy ma ono charakter tauopatyczny. W związku z tym linie podziału CPCS i etapów I i II charakterystyki klinicznej McKee’go nie są w pełni ustalone.
Nie odnotowano wyraźnych predyspozycji genetycznych do CTE. Jednakże gen ApoE4, najbardziej znany czynnik ryzyka dla choroby Alzheimera (25), został powiązany z większymi deficytami poznawczymi i bardziej wydłużonym okresem rekonwalescencji po urazowym uszkodzeniu mózgu (TBI) (11). Badanie przeprowadzone na grupie bokserów wykazało cięższe wyniki u osób posiadających co najmniej jeden allel ApoE4 (26). I odwrotnie, ApoE3 może zapewniać neuroprotekcję, nawet w obecności postępującej patologii CTE (15). Innym proponowanym czynnikiem ochronnym związanym z korzystniejszym powrotem do zdrowia po TBI jest rezerwa poznawcza, mierzona na podstawie przedchorobowego IQ i całkowitej objętości wewnątrzczaszkowej (27). Inni genetyczni kandydaci do dalszych badań obejmują gen białka tau związanego z mikrotubulami (MAPT), gen progranuliny (GRN) i gen otwartej ramki odczytu chromosomu dziewiątego 72 (C9ORF72) (11).
Synergizm patologiczny tauopatii i neurozapalenia jest coraz częściej rozpoznawany. Uważa się, że zewnątrzkomórkowe wydzielanie hiperfosforylowanego tau aktywuje mikroglej i astrocyty, prowadząc do produkcji cytokin prozapalnych, takich jak IL1ß i TNFa, co z kolei prowadzi do aktywacji kinaz tau, takich jak p38 i cdk5, i dalszej fosforylacji tau. Proces ten tworzy błędny, niekończący się cykl tauopatii i neuroinflamacji (28). Biorąc pod uwagę silny związek między powtarzającymi się urazowymi uszkodzeniami mózgu a ryzykiem wystąpienia CTE (1), leczenie TBI w odpowiednim czasie może zmniejszyć rozwój CTE. Prozapalna natura TBI została wcześniej opisana (13), a środki przeciwzapalne, takie jak minocyklina z N-acetylocysteiną, silnym antyoksydantem, podawane w ostrych i podostrych oknach czasowych po TBI, oferują obiecujący schemat terapeutyczny (29, 30). Opracowanie protokołu wrażliwego na czas, przypominającego algorytm leczenia udaru niedokrwiennego, potencjalnie zmierzyłoby długoterminowe wyniki w powrocie do zdrowia po TBI i zapobieganiu rozwojowi późniejszej patologii CTE (29).
Nie ma obecnie leków modyfikujących przebieg choroby CTE, co sprawia, że zapobieganie jest najskuteczniejszym sposobem zwalczania tej wyniszczającej choroby neurodegeneracyjnej (31). Biorąc pod uwagę częstotliwość zderzeń głowami w sportach kontaktowych, takich jak futbol amerykański, zapobieganie urazom głowy będzie wymagało zmiany kulturowej w sposobie nauczania i uprawiania tego sportu. Wymierne korzyści przyniesie szkolenie w zakresie technik bezpiecznego uprawiania sportu, takich jak bezpieczne chwytanie i uderzanie, przy jednoczesnym karaniu lekkomyślnych uderzeń. Dalsze zmiany muszą obejmować tworzenie środowiska bezpieczeństwa, w którym zawodnicy są zachęcani do zgłaszania objawów sędziom, trenerom, a także lekarzom drużyny. Ponadto, ustalenie podstawowego profilu neurokognitywnego mogłoby być wykorzystane jako kliniczny punkt odniesienia do śledzenia zmian w obrazie neuropsychiatrycznym zawodników. Na lekarzach drużyny spoczywa obowiązek usunięcia z boiska zawodników, którzy doznali nawet łagodnego, niepowikłanego TBI, w celu przeprowadzenia dalszej oceny (32).
Istnieje wiele istniejących wyzwań związanych z CTE, którymi należy się zająć. Chociaż częstość występowania wstrząśnienia mózgu związanego ze sportem wynosi od 1,6 mln do 3,8 mln, częstość występowania i rozpowszechnienie CTE pozostają w dużej mierze nieznane (33). Jednym z wyjaśnień tej luki w wiedzy jest być może fakt, że sportowcy narażeni na kumulujące się uderzenia podkorowe, które wywierają wystarczającą siłę do uszkodzenia neuronów, ale początkowo nie dają jawnych objawów klinicznych, często nie są oceniani lub diagnozowani w odpowiednim czasie (34). Prospektywne badania na dużą skalę, takie jak śledzenie sportowców z wieloma TBI w określonym czasie, przyczyniłyby się do lepszego zrozumienia naturalnego przebiegu i fenomenologii choroby. CTE w coraz większym stopniu trafia do opinii publicznej za pośrednictwem środków masowego przekazu. Konieczne są nieustanne wysiłki w celu diagnozowania, oceny i leczenia tej wyniszczającej choroby. Gwałtowny postęp w technikach neuroobrazowania i zrozumienie mechanizmów neuropatologicznych choroby doprowadzi do wcześniejszego rozpoznania i podjęcia interwencji terapeutycznych w odpowiednim czasie.
Wkład autora
Autor potwierdza, że jest jedynym autorem tej pracy i zatwierdził ją do publikacji.
Conflict of Interest Statement
Autor oświadcza, że badania zostały przeprowadzone przy braku jakichkolwiek komercyjnych lub finansowych relacji, które mogłyby być interpretowane jako potencjalny konflikt interesów.
Podziękowania
Autor dziękuje za wielkie wsparcie, wskazówki i mentoring dr. Stephena Strittmattera.
1. McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. (2009) 68:709-35. doi: 10.1097/NEN.0b013e3181a9d503
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, et al. Przewlekła traumatyczna encefalopatia u weteranów wojskowych narażonych na wybuch i model mysi neurotraumy po wybuchu. Sci Transl Med. (2012) 4:134ra60. doi: 10.1126/scitranslmed.3003716
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Martland H. Dementia pugilistica. JAMA. (1928) 91:364-65. doi: 10.1001/jama.1928.02700150029009
CrossRef Full Text | Google Scholar
4. Millspaugh J. Dementia pugilistica. US Naval Med Bull. (1937) 35:303.
Google Scholar
5. Critchley M. Hommage a Clovis Vincent. Paris: Maloine (1949).
Google Scholar
6. Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, et al. Kliniczno-patologiczna ocena przewlekłej encefalopatii urazowej u graczy futbolu amerykańskiego. JAMA. (2017) 318:360-70. doi: 10.1001/jama.2017.8334
PubMed Abstract | CrossRef Full Text | Google Scholar
7. McKee AC, Stein TD, Kiernan PT, Alvarez VE. Neuropatologia przewlekłej encefalopatii pourazowej. Brain Pathol. (2015) 25:350-64. doi: 10.1111/bpa.12248
PubMed Abstract | CrossRef Full Text | Google Scholar
8. McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, et al. Pierwsze spotkanie konsensusu NINDS/NIBIB w celu określenia kryteriów neuropatologicznych dla rozpoznania przewlekłej encefalopatii urazowej. Acta Neuropathol. (2016) 131:75-86. doi: 10.1007/s00401-015-1515-z
PubMed Abstract | CrossRef Full Text | Google Scholar
9. McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. (2013) 136:43-64. doi: 10.1093/brain/aws307
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Jordan BD. Spektrum kliniczne urazowego uszkodzenia mózgu związanego ze sportem. Nat Rev Neurol. (2013) 9:222-30. doi: 10.1038/nrneurol.2013.33
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Stern RA, Daneshvar DH, Baugh CM, Seichepine DR, Montenigro PH, Riley DO, et al. Prezentacja kliniczna przewlekłej encefalopatii pourazowej. Neurology. (2013) 81:1122-9. doi: 10.1212/WNL.0b013e3182a55f7f
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Gardner A, Iverson GL, McCrory P. Chronic traumatic encephalopathy in sport: a systematic review. Br J Sports Med. (2014) 48:84-90. doi: 10.1136/bjsports-2013-092646
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Montenigro PH, Baugh CM, Daneshvar DH, Mez J, Budson AE, Au R, et al. Podtypy kliniczne przewlekłej encefalopatii urazowej: przegląd literatury i proponowane kryteria diagnostyczne badań dla zespołu encefalopatii urazowej. Alzheim Res Ther. (2014) 6:68. doi: 10.1186/s13195-014-0068-z
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Mawdsley C, Ferguson F. Neurological disease in boxers. Lancet. (1963) 282:795-801. doi: 10.1016/S0140-6736(63)90498-7
CrossRef Full Text | Google Scholar
15. Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery. (2005) 57:128-34. doi: 10.1227/01.NEU.0000163407.92769.ED
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. (2011) 134:2456-77. doi: 10.1093/brain/awr179
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Omalu BI, Bailes J, Hammers JL, Fitzsimmons RP. Przewlekła encefalopatia pourazowa, samobójstwa i paraseryjność u zawodowych sportowców amerykańskich: rola patologa sądowego. Am J Forensic Med Pathol. (2010) 31:130-2. doi: 10.1097/PAF.0b013e3181ca7f35
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Maroon JC, Winkelman R, Bost J, Amos A, Mathyssek C, Miele V. Chronic traumatic encephalopathy in contact sports: a systematic review of all reported pathological cases. PLoS ONE. (2015) 10:e0117338. doi: 10.1371/journal.pone.0117338
CrossRef Full Text | Google Scholar
19. Iverson GL. Przewlekła encefalopatia traumatyczna i ryzyko samobójstwa u byłych sportowców. Br J Sports Med. (2014) 48:162-4. doi: 10.1136/bjsports-2013-092935
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Iverson GL, Gardner AJ, McCrory P, Zafonte R, Castellani RJ.Iverson A critical review of chronic traumatic encephalopathy. Neurosci Biobehav Rev. (2015) 56:276-93. doi: 10.1016/j.neubiorev.2015.05.008
CrossRef Full Text | Google Scholar
21. Randolph C. Przewlekła encefalopatia traumatyczna nie jest prawdziwą chorobą. Arch Clin Neuropsychol. (2018) 33:644-8. doi: 10.1093/arclin/acy063
CrossRef Full Text | Google Scholar
22. Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R, et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature. (2019) 568:420-3. doi: 10.1038/s41586-019-1026-5
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Weir DR, Jackson JS, Sonnega A, et al. National Football League Player Care Foundation Study of Retired NFL Players. Ann Arbor: University of Michigan Institute for Social Research (2009).
Google Scholar
24. King NS, Kirwilliam S. Permanent post-concussion symptoms after mild head injury. Brain Inj. (2011) 25:462-70. doi: 10.3109/02699052.2011.558042
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Ward A, Crean S, Mercaldi CJ, Collins JM, Boyd D, Cook MN, et al. Częstość występowania genotypu i homozygot apolipoproteiny E4 (APOE e4/4) wśród pacjentów, u których zdiagnozowano chorobę Alzheimera: przegląd systematyczny i metaanaliza. Neuroepidemiology. (2012) 38:1-17. doi: 10.1159/000334607
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Jordan BD, Relkin NR, Ravdin LD, Jacobs AR, Bennett A, Gandy S. Apolipoproteina Eε4 związana z przewlekłym urazowym uszkodzeniem mózgu w boksie. (1997) 278:136-40. doi: 10.1001/jama.278.2.136
CrossRef Full Text | Google Scholar
27. Stern Y. Rezerwa poznawcza w starzeniu się i chorobie Alzheimera. Lancet Neurol. (2012) 11:1006-12. doi: 10.1016/S1474-4422(12)70191-6
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Laurent CL, Buée L, Blum D. Tau i neuroinflammation: Jaki wpływ na chorobę Alzheimera i tauopatie? Biomed J. (2018) 41:21-33. doi: 10.1016/j.bj.2018.01.003
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Sangobowale MA, Grin’kina NM, Whitney K, Nikulina E, St., Laurent-Ariot K, Ho JS, et al. Minocyklina plus N-acetylocysteina zmniejszają deficyty behawioralne i poprawiają histologię z klinicznie użytecznym oknem czasowym. J Neurotrauma. (2018) 35:907-17. doi: 10.1089/neu.2017.5348
CrossRef Full Text | Google Scholar
30. Haber M, James J, Kim J, Sangobowale M, Irizarry R, Ho J, et al. Minocyklina plus N-acteylcysteina indukuje remielinizację, synergicznie chroni oligodendrocyty i modyfikuje neuroinflammation w szczurzym modelu łagodnego urazowego uszkodzenia mózgu. J Cereb Blood Flow Metab. (2018) 38:1312-6. doi: 10.1177/0271678X17718106
PubMed Abstract | CrossRef Full Text | Google Scholar
31. Livingston G, Sommerlad A, Orgeta V, Costafreda SG, Huntley J, Ames D, et al. Dementia prevention, intervention and care. Lancet. (2017) 390: 2673-734. doi: 10.1016/S0140-6736(17)31363-6
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Saulle M, Greenwald BD. Przewlekła encefalopatia pourazowa: przegląd. Rehabil Res Pract. (2012). 2012: 816069. doi: 10.1155/2012/816069
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Langlois JA, Rutland-Brown W, Wald MM. Epidemiologia i wpływ traumatycznego urazu mózgu. J Head Trauma Rehabil. (2006) 21:375-8. doi: 10.1097/00001199-200609000-00001
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Martini D, Eckner J, Kutcher J, Broglio SP. Subconcussive head impact biomechanics: comparing differing offensive schemes. Med Sci Sports Exerc. (2013) 45:755-61. doi: 10.1249/MSS.0b013e3182798758
PubMed Abstract | CrossRef Full Text | Google Scholar
.