Key Words: progressive hemifacial atrophy, Parry-Romberg syndrome
Progressive hemifacial atrophy- or Parry-Romberg syndrome- is a rare disease, classified as one of the forms of localized morphea or scleroderma. A sua causa é desconhecida. Caracteriza-se pela atrofia da pele, gordura, músculos e estruturas osteocartilaginosas subjacentes que geralmente afetam unilateralmente a face e o pescoço, e está associada a sintomas neurológicos (epilepsia secundária) e envolvimento de outros órgãos e sistemas. O seu curso é lento e progressivo e começa nas primeiras duas décadas de vida. A predileção pelo sexo feminino tem sido observada. Relatamos o caso de uma menina de 10 anos diagnosticada no Hospital Hipólito Unánue em Tacna, Peru. O conhecimento desta condição é importante no diagnóstico diferencial de morfemas localizadas ou esclerodermia.
Saber ideias-chave
- Atrofia hemifacial progressiva é uma doença rara, com uma incidência de três casos por 100 000 pessoas por ano; é mais frequente nas mulheres e a sua etiologia é desconhecida.
- Caracteriza-se pela progressão da atrofia, geralmente unilateral no lado esquerdo da face, o que compromete as estruturas osseocartilaginosas.
- No caso relatado, o paciente apresentou a lesão do lado direito, achado pouco freqüente.
- O conhecimento desta doença é importante para o diagnóstico diferencial de morfema ou esclerodermia localizada.
Introdução
Atrofia hemifacial progressiva, também conhecida como síndrome de Parry-Romberg, foi descrita pela primeira vez por Caleb Hillier Parry e Moritiz Heinrich Romberg, em 1825 e 1846, e caracteriza-se por atrofia progressiva da pele e tecidos subjacentes, afetando a face e o pescoço, geralmente de forma unilateral. A sua incidência é de cerca de três casos por 100 000 pessoas por ano. Embora esta doença rara normalmente só afecte a face e o pescoço unilateralmente, ocasionalmente manifesta-se bilateralmente e afecta até o tronco, braços e pernas. Seu curso é progressivo, lento e surge principalmente entre os dois e 20 anos de idade,.
A patogênese desta doença é desconhecida; no entanto, uma variedade de teorias tem sido proposta, incluindo causas imunológicas, traumáticas, infecciosas, endocrinológicas, neurológicas e genéticas. A epilepsia é a anomalia mais frequente associada à síndrome de Perry-Romberg.
Nós relatamos uma menina de 10 anos de idade diagnosticada com atrofia hemifacial progressiva no Hospital Hipolito Unanue em Tacna, Peru, em 2015. Até onde sabemos, este é o terceiro caso relatado no Peru e o primeiro caso em Tacna.
Relatório de caso
Este é o caso de uma paciente de 10 anos de idade de Tacna (uma cidade no sul do Peru que faz fronteira com a Bolívia e o Chile). Ela é o produto de uma segunda gestação e nasceu a termo por parto cesáreo. Seus pais não são parentes de sangue e seus familiares imediatos são presumivelmente saudáveis. No período perinatal, não foi detectada nenhuma doença relevante. O desenvolvimento físico e motor foi adequado.
Nossa paciente chegou ao serviço de emergência do Hospital Hipólito Unánue apresentando perda de consciência e convulsões tónico-clónicas que evoluíram para o estado epiléptico focal. Por este motivo, a paciente foi internada para estudos e tratamento posterior.
Ao exame físico, observamos a presença de duas lesões atróficas na face: a primeira e mais longa lesão, localizada na linha medial facial, tinha oito centímetros de comprimento, dois centímetros de largura e entre um e dois milímetros de profundidade, estendendo-se do tríceps até a asa direita do nariz; notou-se leve hiperpigmentação no seu ápice (Figuras 1 e 2, indicadas pela seta vermelha). A segunda lesão atrófica, localizada lateralmente a partir da primeira lesão, estendeu-se da linha frontal direita até o arco supraciliar direito, onde foi observada uma zona de madarose, com quatro centímetros de comprimento, um centímetro de largura e sete milímetros de profundidade (Figuras 1 e 2, lesão indicada pela seta verde).
Figure 1. Lesões atróficas na face.
Figura 2. Zona de madarose.
Na região parietal direita, foi encontrada uma zona de alopecia (Figura 3). Ao mesmo tempo, observamos anisocoria e midríase da pupila do olho direito, mas ainda era foto-reativa. Também foram observadas ptose palpebral direita moderada, úlcera da córnea direita, luxação do cristalino direito e ametropia. Não foram encontradas anomalias na retina. Atrofia da asa direita do nariz foi notada, juntamente com desvio do septo, comissura oral direita elevada, acentuação do sulco nasogeniano direito (Figura 2, seta amarela) e posição anormal dos dentes (Figura 4). O restante do exame foi normal.
Figure 3. Zona alopécica na região parietal direita.
Figure 4. Posição anormal dos dentes.
A partir dos três anos de idade, nosso paciente sofreu traumatismo cerebral com perda de consciência e convulsões. Naquela época, esses sintomas foram assumidos como sendo causados pelo trauma. Um ano depois, seus pais notaram a presença e progressão da anormalidade da face (descrita nos parágrafos acima); entretanto, não procuraram assistência médica, pois assumiram ser conseqüência do trauma anterior.
Os exames laboratoriais que realizamos para a paciente apresentaram leve leucocitose com neutrofilia (18,3 x 103 células por mm3, 79% neutrófilos) e glicemia elevada (149,8mg/dl). Estes achados foram explicados por convulsões. Obtivemos resultados negativos (normais) para teste de anticorpos anti-nucleares, anticorpos citoplasmáticos anti-neutrófilos, antígeno de superfície da hepatite B e IgM contra o vírus da hepatite A.
A tomografia computadorizada (TC) do cérebro mostrou calcificações e edema circundante nos gânglios basais direitos: no putamen e na cabeça do núcleo do caudato (Figura 5). Na reconstrução 3D tomográfica do crânio, encontramos assimetria dos ossos faciais: atrofia do osso maxilar direito e do osso frontal, posicionamento anormal dos dentes, desvio do septo nasal e crescimento excessivo do turbinado inferior esquerdo (Figura 6).
Figure 5. Calcificações nos gânglios basais do cérebro.
Figure 6. Desvio do septo nasal com convexidade esquerda.
Após a obtenção dos dados da anamnese, exame físico e testes diagnósticos, o autor principal deste artigo realizou uma pesquisa exaustiva em bases de dados de doenças raras (como Mendelian Inheritance Online in Man – OMIM, disponível em www.omim.org), concluindo que a condição da menina era compatível com a síndrome de Parry-Romberg ou atrofia hemifacial progressiva.
A paciente obteve controle adequado das convulsões, recebeu alta do hospital, porém está sob observação médica de um neurologista, pediatra, oftalmologista e psicólogo. Esta equipe multidisciplinar tem o objetivo de melhorar a qualidade de vida da nossa paciente.
Discussão
Síndrome de Parry-Romberg é uma doença rara caracterizada pela atrofia progressiva da pele, músculos e ossos que compromete a face, geralmente unilateralmente, e em alguns casos é acompanhada por anormalidade da dentição, língua e palato. Embora a síndrome de Parry-Romberg seja geralmente unilateral, é possível encontrar pacientes com danos bilaterais, mesmo encontrando aqueles com tronco, braços e pernas comprometidos.
A incidência desta doença é de três casos por 100 000 pacientes por ano e é mais comum em mulheres com proporção de 3:1. Normalmente se apresenta nos primeiros vinte anos de vida.
Esta doença é geralmente classificada como esclerodermia linear localizada ou morfema. Na literatura mundial, encontramos referências à síndrome de Parry-Romberg como morfema em golpe de sabre (“golpe de sabre”, devido à similitude da atrofia da pele à ferida causada pelo sabre). No entanto, do nosso entendimento, estas condições são duas entidades diferentes, pois os pacientes que sofrem da síndrome de Parry-Romberg não apresentam características histológicas da esclerose dérmica, enquanto que os pacientes com morfema no golpe de sabre o apresentam,.
A etiologia da síndrome de Parry-Romberg ainda é desconhecida; no entanto, a hipótese mais aceita é a etiologia auto-imune,. As vias inflamatórias Th1 e Th17 são de grande importância na fisiopatologia desta doença, pelo menos nos estágios iniciais da mesma. A via inflamatória Th2 é importante no estágio tardio, o estágio fibrótico. Esta resposta inflamatória dos tecidos pode ser iniciada por um evento traumático incidental, cirurgia, ou mesmo por um trauma obstétrico. A teoria neurológica sugere que esta doença é o resultado da migração desorganizada das células da crista neural que compromete um ou mais ramos do nervo trigêmeo.
Alguns relatos afirmam que a síndrome de Parry-Romberg é uma doença genética autossômica dominante. Além disso, tem sido dito que a atrofia hemifacial progressiva está ligada à doença de Lyme, Borrelia burgdorferi, e a outras infecções como sífilis, rubéola, tuberculose e o vírus da hepatite B.
Neste caso, o paciente desenvolveu a doença após uma queda, adquirindo traumatismo craniano. Não havia registros prévios de doença na família. Seus exames laboratoriais para presença de anticorpos anti-nucleares e hepatite viral foram negativos. Não avaliamos nossa paciente para a doença de Lyme porque o Peru não é uma área endêmica para esta doença, e ela negou ter viajado para regiões endêmicas.
A síndrome de Parry-Romberg envolve dermatomas de um ou mais ramos do nervo trigêmeo e geralmente compromete a metade esquerda da face,. Pode apresentar algum grau de aumento de pigmentação e luminosidade da pele. Alopecia pode ser vista em qualquer área do couro cabeludo, mas geralmente na região frontoparietal,.
M. Wong, em sua série de casos, demonstra uma predileção pela metade esquerda da face. Carlos Galarza, em seus dois casos relatados de Cuzco, Peru, também demonstrou atrofia da pele do lado esquerdo. Entretanto, nosso paciente apresentou atrofia cutânea localizada na metade direita da face, que se estendeu das regiões frontal e mandibular, acompanhada de leve hiperpigmentação, desvio de septo, distribuição anormal dos dentes e localização parietal da alopecia.
As manifestações oculares da síndrome de Parry-Romberg podem estar presentes antes, durante ou após o aparecimento da atrofia cutânea, e podem incluir: enofthalmos levando ao diplópode (causado pela atrofia de gordura no espaço retro bulbar), olho seco, ou uveíte. Também tem havido relatos de pigmentação das pálpebras, fotofobia, estrabismo, atrofia da íris, panuveite, corpo ciliar hipotônico, vasculite retiniana, edema e descolamento da retina, etc.
A avaliação neurológica pode mostrar paralisia do nervo oculomotor manifestada com anormalidades pupilares, anisocoria, midríase ou miose no lado afetado e envolvimento do nervo óptico que pode levar a neuroretinite e papilite. Nossa paciente, desde sua primeira visita ao departamento de emergência, sofreu anisocoria grave e midríase do olho direito, uma anormalidade da reação pupilar, e à medida que a doença progredia, outras manifestações apareceram: deslocamento do cristalino direito, úlcera corneana, enofthalmos grave e ametropia.
Dalla Costa, em sua revisão mundial de 205 pacientes diagnosticados com síndrome de Parry-Romberg, relatou que 50% dos pacientes apresentavam sintomas de comprometimento do sistema nervoso central e 15% tinham crises epiléticas, dores de cabeça, dor facial, comprometimento dos nervos cranianos e hemiplegia.
A manifestação neurológica mais frequente da síndrome de Parry-Romberg é a epilepsia (60,5% de freqüência). As convulsões focais ipsilateralmente às calcificações cerebrais são comuns (observadas em 50% do total de casos). Com uma frequência de 33%, a epilepsia secundária é muito difícil de tratar. Dor de cabeça (44%) e neuralgia do trigêmeo (8,5%) também podem estar presentes.
Calcificações intraparenquimatosas do cérebro do mesmo lado das anormalidades faciais são os achados mais frequentes em tomografia computadorizada e ressonância magnética. Hiperintensidades da matéria branca, infração focal do corpo caloso, hemiatrofia cerebral e realce leptomeníngeo, aneurismas intracranianos e malformações vasculares e microhemorragia cerebral têm sido descritos. Não há correlação entre neuroimagem e características faciais desta doença.
As características neurológicas e oculares são as manifestações mais frequentes da síndrome de Parry-Romberg, é possível encontrar anormalidades cardíacas (cardiomiopatia hipertrófica), anormalidades endócrinas (hipertiroidismo, hipotiroidismo), doenças auto-imunes (cirrose biliar primária, artrite reumatóide, esclerose múltipla), e doenças congênitas, como a síndrome de Poland, microftalmia e malformações renais.
Epilepsia secundária e dores de cabeça foram as características neurológicas predominantes no nosso caso. As convulsões epilépticas tinham sido bem controladas. Tomografia e ressonância foram descritas como calcificações em gânglios basais: putamen e caudato do núcleo. Não encontramos nenhuma outra anormalidade além das descritas acima.
O objetivo do tratamento é parar a fase ativa da doença. Isto pode ser conseguido com a prescrição de metotrexato a 0,3 – 1mg/kg/semana durante um a dois anos, e pode ser associado com prednisona durante os primeiros três meses considerando o efeito retardado do metotrexato na inflamação e fibrose.
Síndrome de Parry-Romberg tem um grande impacto biopsicossocial na vida do paciente devido a limitações funcionais e estéticas. Algumas abordagens cirúrgicas para restaurar a simetria facial têm sido propostas. Casos leves e moderados podem ser tratados com preenchimentos de silicone, colágeno, implantes porosos de polietileno e enxerto autólogo de gordura. A enxertia de cartilagem, ossos, gordura e pele são opções de tratamento para casos mais graves. O objetivo do tratamento psicológico é a reintegração social dos pacientes.
O prognóstico da doença depende da idade do paciente, sendo mais grave quando as alterações atróficas começam na idade adulta (20 anos de idade ou mais). A gravidade da atrofia, dano cerebral e má resposta ao tratamento são maiores em pacientes com mais de 20 anos de idade. Esses determinantes nos dão a abordagem global das seqüelas e o tratamento que deve ser considerado para se obter melhores resultados terapêuticos.
Conclusões
Hemi atrofia facial progressiva, ou síndrome de Parry-Romberg, é uma doença rara, progressiva, de causa desconhecida, que afeta principalmente as mulheres. Há poucos casos relatados desta síndrome no Peru, sendo este o primeiro caso na região de Tacna.
Caracteriza-se pela progressão da atrofia, geralmente unilateral, sendo mais frequente no lado esquerdo da face e envolvendo estruturas osteocartilaginosas. No nosso caso, a anormalidade cutânea foi encontrada no lado direito; este é um achado pouco freqüente.
O conhecimento desta doença torna-se relevante no diagnóstico diferencial de lesões atróficas em um lado da face. Dentre as doenças com as quais o diagnóstico diferencial deve ser feito, destacam-se: doença do golpe de sabre, lúpus profundo, atrofia de esteróides e outros tipos de morfemas.
Contribuições do autor
EOL: autor principal, diagnóstico e acompanhamento do paciente, redação e revisão crítica do artigo, aprovação final do artigo e assunção de responsabilidade pelo manuscrito. SDA: autor, redação e contribuições importantes para o artigo e assunção de responsabilidade pelo manuscrito. PDA: co-autor, contribuições importantes para o artigo
Conflito de interesses
Nenhum dos autores declara ter conflito de interesses com o assunto deste artigo.
Fonte de financiamento
O financiamento na elaboração do artigo é próprio.
Aspectos éticos
Os autores obtiveram o consentimento informado dos pais da paciente (sendo ela menor de idade) para a publicação deste artigo e das imagens que o acompanham.
Dados
Declaramos disponibilidade para a entrega dos dados mediante solicitação.
Dos editores
A versão original deste manuscrito foi submetida em espanhol. Esta versão em inglês foi submetida pelos autores e foi ligeiramente editada pela Revista.

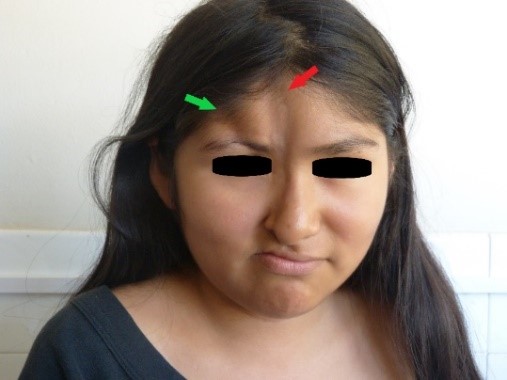 Figure 1. Lesões atróficas na face.
Figure 1. Lesões atróficas na face. 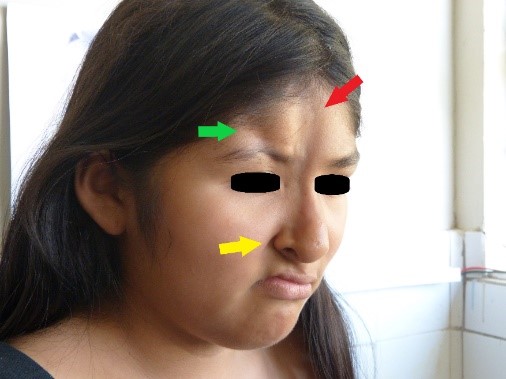 Figura 2. Zona de madarose.
Figura 2. Zona de madarose. 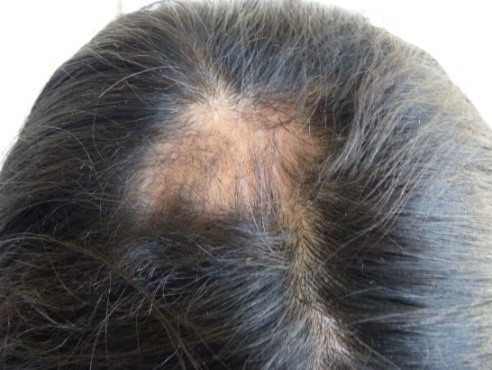 Figura 3. Zona alopécica na região parietal direita.
Figura 3. Zona alopécica na região parietal direita. 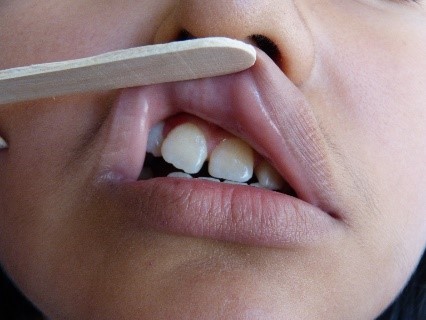 Figura 4. Posição anormal dos dentes.
Figura 4. Posição anormal dos dentes. 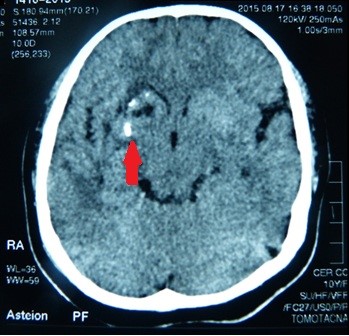 Figura 5. Calcificações nos gânglios basais do cérebro.
Figura 5. Calcificações nos gânglios basais do cérebro. 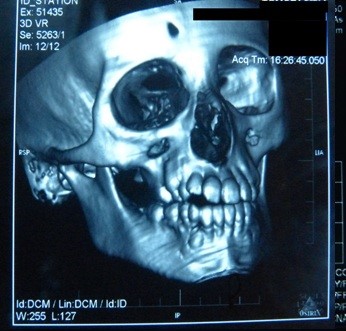 Figura 6. Desvio do septo nasal com convexidade esquerda.
Figura 6. Desvio do septo nasal com convexidade esquerda.